

G551D and G1349D, two CF-associated mutations in the signature sequences of CFTR, exhibit distinct gating defects
- PMID: 17353351
- PMCID: PMC2151620
- DOI: 10.1085/jgp.200609667
G551D and G1349D, two CF-associated mutations in the signature sequences of CFTR, exhibit distinct gating defects
Abstract
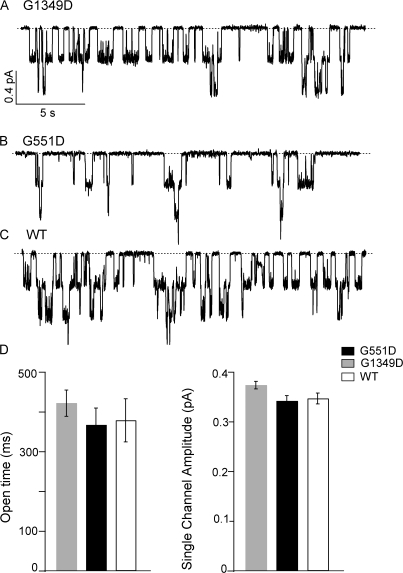
Mutations in the gene encoding cystic fibrosis transmembrane conductance regulator (CFTR) result in cystic fibrosis (CF). CFTR is a chloride channel that is regulated by phosphorylation and gated by ATP binding and hydrolysis at its nucleotide binding domains (NBDs). G551D-CFTR, the third most common CF-associated mutation, has been characterized as having a lower open probability (Po) than wild-type (WT) channels. Patients carrying the G551D mutation present a severe clinical phenotype. On the other hand, G1349D, also a mutant with gating dysfunction, is associated with a milder clinical phenotype. Residues G551 and G1349 are located at equivalent positions in the highly conserved signature sequence of each NBD. The physiological importance of these residues lies in the fact that the signature sequence of one NBD and the Walker A and B motifs from the other NBD form the ATP-binding pocket (ABP1 and ABP2, named after the location of the Walker A motif) once the two NBDs dimerize. Our studies show distinct gating characteristics for these mutants. The G551D mutation completely eliminates the ability of ATP to increase the channel activity, and the observed activity is approximately 100-fold smaller than WT-CFTR. G551D-CFTR does not respond to ADP, AMP-PNP, or changes in [Mg(2+)]. The low activity of G551D-CFTR likely represents the rare ATP-independent gating events seen with WT channels long after the removal of ATP. G1349D-CFTR maintains ATP dependence, albeit with a Po approximately 10-fold lower than WT. Interestingly, compared to WT results, the ATP dose-response relationship of G1349D-CFTR is less steep and shows a higher apparent affinity for ATP. G1349D data could be well described by a gating model that predicts that binding of ATP at ABP1 hinders channel opening. Thus, our data provide a quantitative explanation at the single-channel level for different phenotypes presented by patients carrying these two mutations. In addition, these results support the idea that CFTR's two ABPs play distinct functional roles in gating.
Figures










References
-
- Ai, T., S.G. Bompadre, X. Wang, S. Hu, M. Li, and T.-C. Hwang. 2004. Capsaicin potentiates wild-type and mutant CFTR chloride channel currents. Mol. Pharmacol. 65:1415–1426. - PubMed
-
- Aleksandrov, L., A. Mengos, X.-B. Chang, A. Aleksandrov, and J.R. Riordan. 2001. Differential interactions of nucleotides at the two nucleotide binding domains of the cystic fibrosis transmembrane conductance regulator. J. Biol. Chem. 276:12918–12923. - PubMed
-
- Anderson, M.P., and M.J. Welsh. 1992. Regulation by ATP and ADP of CFTR chloride channels that contain mutant nucleotide-binding domains. Science. 257:1701–1704. - PubMed
-
- Beaudet, A.L., G.L. Feldman, K. Kobayashi, W.K. Lemma, S.D. Fernbach, M.R. Knowles, R.C. Boucher, and W.E. O'Brien. 1991. Mutation analysis for cystic fibrosis in a North American population. In The Identification of the CF (cystic fibrosis) Gene-recent Progress and New Research Strategies. L-C. Tsui, G. Romeo, R. Greger, and S. Gorini, editors. Plenum Press, New York. 53–54. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
