

The Sorcerer II Global Ocean Sampling expedition: expanding the universe of protein families
- PMID: 17355171
- PMCID: PMC1821046
- DOI: 10.1371/journal.pbio.0050016
The Sorcerer II Global Ocean Sampling expedition: expanding the universe of protein families
Abstract
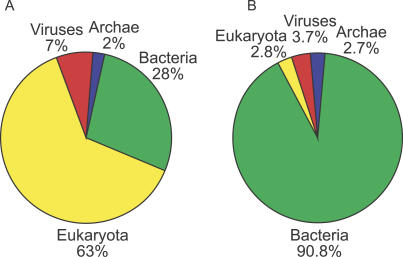
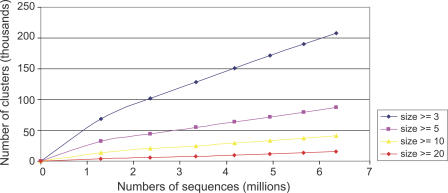
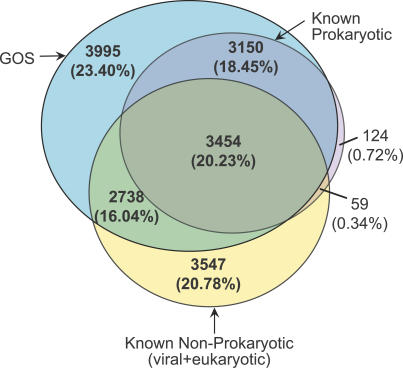
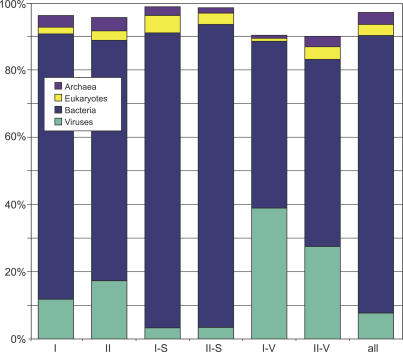
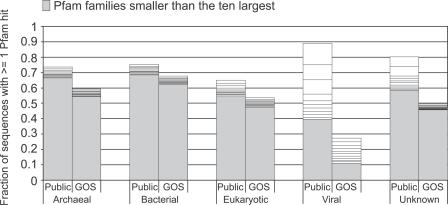
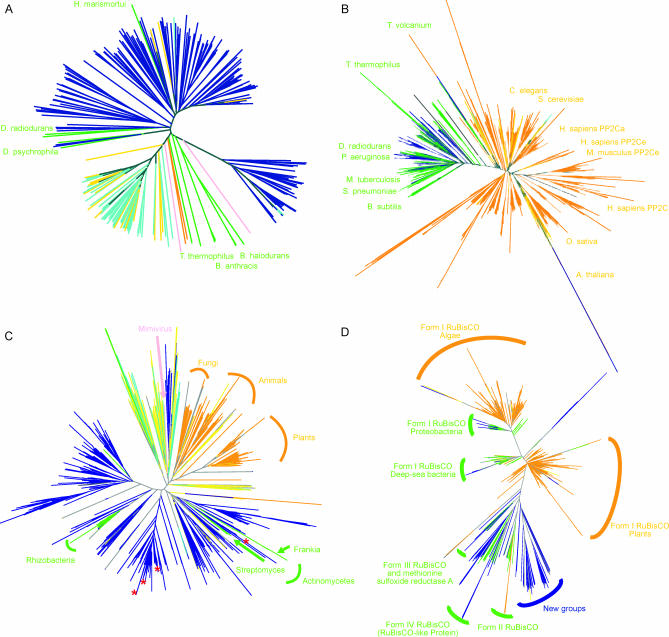
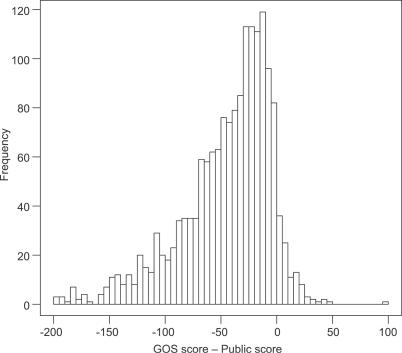
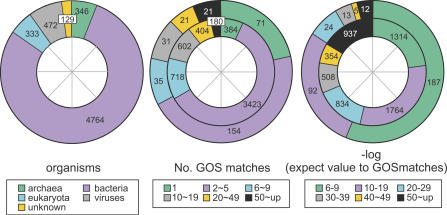
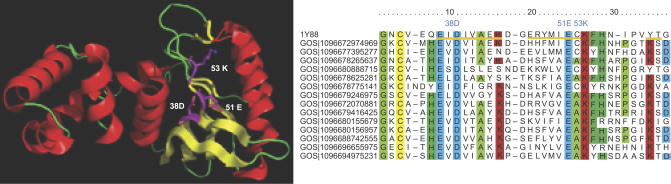
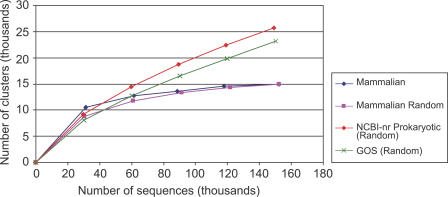
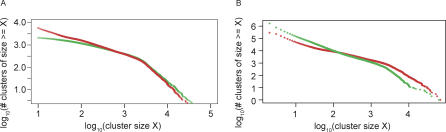
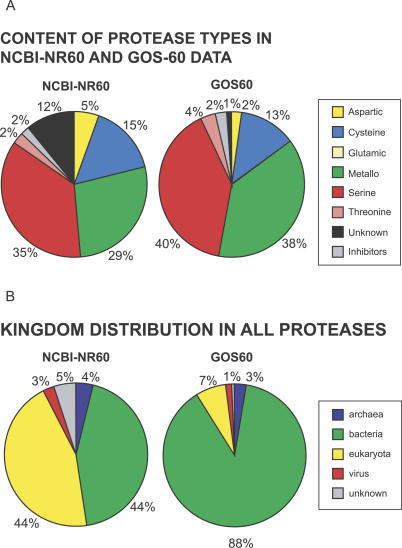
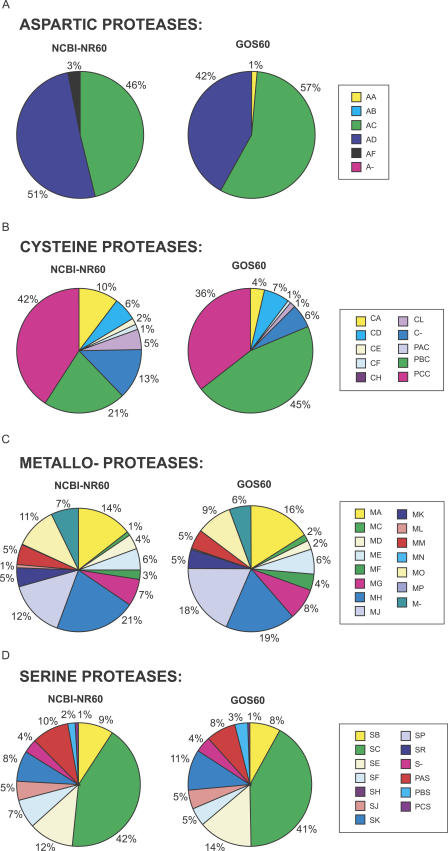
Metagenomics projects based on shotgun sequencing of populations of micro-organisms yield insight into protein families. We used sequence similarity clustering to explore proteins with a comprehensive dataset consisting of sequences from available databases together with 6.12 million proteins predicted from an assembly of 7.7 million Global Ocean Sampling (GOS) sequences. The GOS dataset covers nearly all known prokaryotic protein families. A total of 3,995 medium- and large-sized clusters consisting of only GOS sequences are identified, out of which 1,700 have no detectable homology to known families. The GOS-only clusters contain a higher than expected proportion of sequences of viral origin, thus reflecting a poor sampling of viral diversity until now. Protein domain distributions in the GOS dataset and current protein databases show distinct biases. Several protein domains that were previously categorized as kingdom specific are shown to have GOS examples in other kingdoms. About 6,000 sequences (ORFans) from the literature that heretofore lacked similarity to known proteins have matches in the GOS data. The GOS dataset is also used to improve remote homology detection. Overall, besides nearly doubling the number of current proteins, the predicted GOS proteins also add a great deal of diversity to known protein families and shed light on their evolution. These observations are illustrated using several protein families, including phosphatases, proteases, ultraviolet-irradiation DNA damage repair enzymes, glutamine synthetase, and RuBisCO. The diversity added by GOS data has implications for choosing targets for experimental structure characterization as part of structural genomics efforts. Our analysis indicates that new families are being discovered at a rate that is linear or almost linear with the addition of new sequences, implying that we are still far from discovering all protein families in nature.
Conflict of interest statement
Figures

Comment in
-
Global ocean sampling collection.PLoS Biol. 2007 Mar;5(3):e83. doi: 10.1371/journal.pbio.0050083. PLoS Biol. 2007. PMID: 17355178 Free PMC article.
-
Untapped bounty: sampling the seas to survey microbial biodiversity.PLoS Biol. 2007 Mar;5(3):e85. doi: 10.1371/journal.pbio.0050085. Epub 2007 Mar 13. PLoS Biol. 2007. PMID: 20076663 Free PMC article. No abstract available.
References
-
- Murzin AG, Brenner SE, Hubbard T, Chothia C. SCOP: A structural classification of proteins database for the investigation of sequences and structures. J Mol Biol. 1995;247:536–540. - PubMed
-
- Orengo CA, Michie AD, Jones S, Jones DT, Swindells MB, et al. CATH—A hierarchic classification of protein domain structures. Structure. 1997;5:1093–1108. - PubMed
-
- Thornton JM, Orengo CA, Todd AE, Pearl FM. Protein folds, functions and evolution. J Mol Biol. 1999;293:333–342. - PubMed
-
- Todd AE, Orengo CA, Thornton JM. Evolution of function in protein superfamilies, from a structural perspective. J Mol Biol. 2001;307:1113–1143. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
