

Most rare missense alleles are deleterious in humans: implications for complex disease and association studies
- PMID: 17357078
- PMCID: PMC1852724
- DOI: 10.1086/513473
Most rare missense alleles are deleterious in humans: implications for complex disease and association studies
Abstract
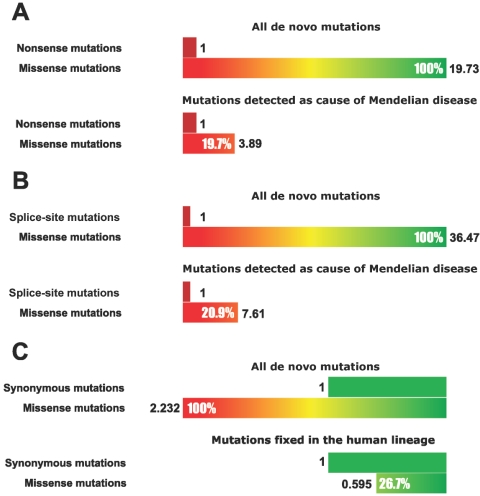
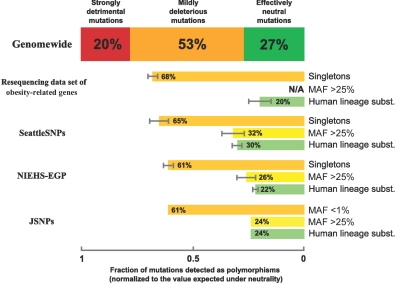
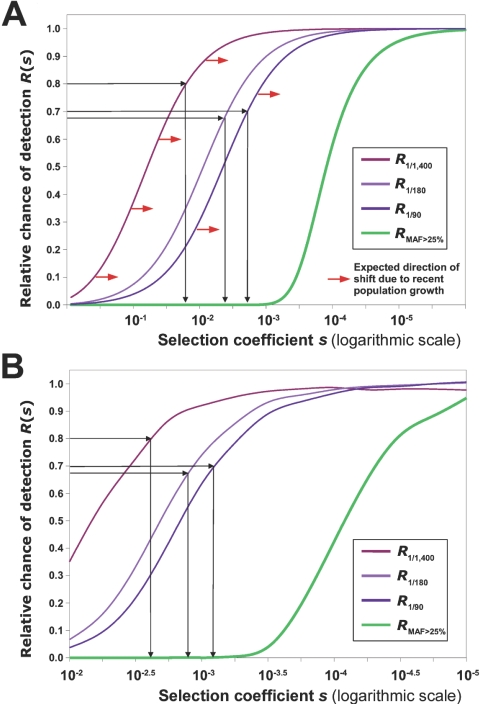
The accumulation of mildly deleterious missense mutations in individual human genomes has been proposed to be a genetic basis for complex diseases. The plausibility of this hypothesis depends on quantitative estimates of the prevalence of mildly deleterious de novo mutations and polymorphic variants in humans and on the intensity of selective pressure against them. We combined analysis of mutations causing human Mendelian diseases, of human-chimpanzee divergence, and of systematic data on human genetic variation and found that ~20% of new missense mutations in humans result in a loss of function, whereas ~27% are effectively neutral. Thus, the remaining 53% of new missense mutations have mildly deleterious effects. These mutations give rise to many low-frequency deleterious allelic variants in the human population, as is evident from a new data set of 37 genes sequenced in >1,500 individual human chromosomes. Surprisingly, up to 70% of low-frequency missense alleles are mildly deleterious and are associated with a heterozygous fitness loss in the range 0.001-0.003. Thus, the low allele frequency of an amino acid variant can, by itself, serve as a predictor of its functional significance. Several recent studies have reported a significant excess of rare missense variants in candidate genes or pathways in individuals with extreme values of quantitative phenotypes. These studies would be unlikely to yield results if most rare variants were neutral or if rare variants were not a significant contributor to the genetic component of phenotypic inheritance. Our results provide a justification for these types of candidate-gene (pathway) association studies and imply that mutation-selection balance may be a feasible evolutionary mechanism underlying some common diseases.
Figures
References
Web Resources
-
- Consensus CDS (CCDS) Project, http://www.ncbi.nlm.nih.gov/CCDS/
-
- Human Gene Mutation Database (HGMD), http://www.hgmd.cf.ac.uk/ac/index.php
-
- JSNP, http://snp.ims.u-tokyo.ac.jp/ (for database of Japanese SNPs)
-
- NIEHS SNPs Program, http://egp.gs.washington.edu/
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
