

Incorporating indel information into phylogeny estimation for rapidly emerging pathogens
- PMID: 17359539
- PMCID: PMC1853084
- DOI: 10.1186/1471-2148-7-40
Incorporating indel information into phylogeny estimation for rapidly emerging pathogens
Abstract
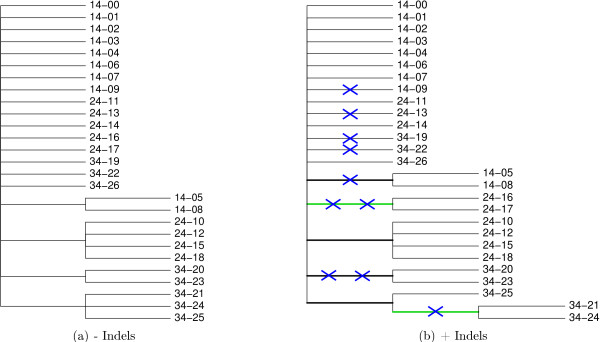
Background: Phylogenies of rapidly evolving pathogens can be difficult to resolve because of the small number of substitutions that accumulate in the short times since divergence. To improve resolution of such phylogenies we propose using insertion and deletion (indel) information in addition to substitution information. We accomplish this through joint estimation of alignment and phylogeny in a Bayesian framework, drawing inference using Markov chain Monte Carlo. Joint estimation of alignment and phylogeny sidesteps biases that stem from conditioning on a single alignment by taking into account the ensemble of near-optimal alignments.
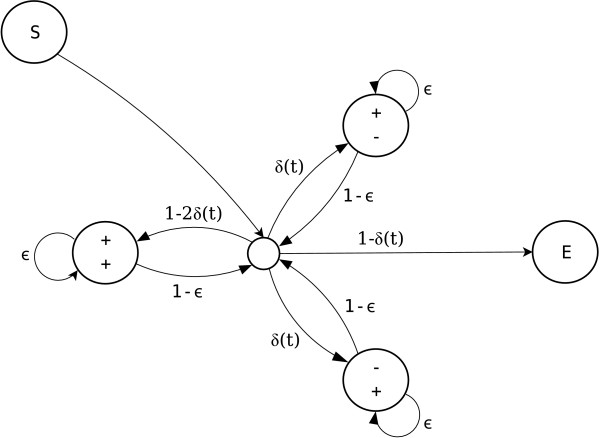
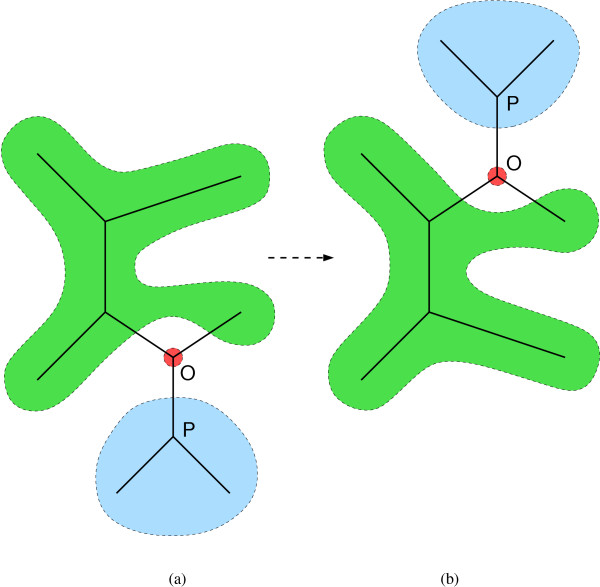
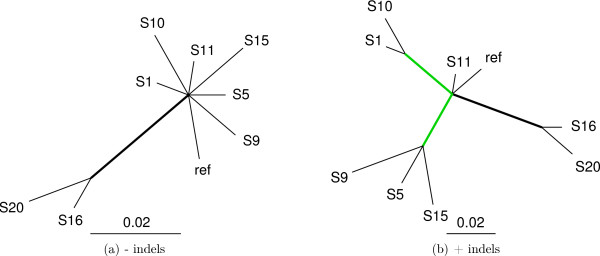
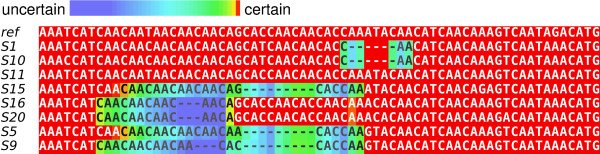
Results: We introduce a novel Markov chain transition kernel that improves computational efficiency by proposing non-local topology rearrangements and by block sampling alignment and topology parameters. In addition, we extend our previous indel model to increase biological realism by placing indels preferentially on longer branches. We demonstrate the ability of indel information to increase phylogenetic resolution in examples drawn from within-host viral sequence samples. We also demonstrate the importance of taking alignment uncertainty into account when using such information. Finally, we show that codon-based substitution models can significantly affect alignment quality and phylogenetic inference by unrealistically forcing indels to begin and end between codons.
Conclusion: These results indicate that indel information can improve phylogenetic resolution of recently diverged pathogens and that alignment uncertainty should be considered in such analyses.
Figures

References
-
- Shankarappa R, Margolick JB, Gange SJ, Rodrigo AG, Upchurch D, Farzadegan h, Gupta P, Rinaldo CR, Learn GH, He X, Huang XL, Mullins JI. Consistent Viral Evolutionary Changes Associated with the Progression of Human Immunodeficiency Virus Type 1 Infection. Journal of Virology. 1999;73:10489–10502. - PMC - PubMed
