

A porphomethene inhibitor of uroporphyrinogen decarboxylase causes porphyria cutanea tarda
- PMID: 17360334
- PMCID: PMC1820519
- DOI: 10.1073/pnas.0700547104
A porphomethene inhibitor of uroporphyrinogen decarboxylase causes porphyria cutanea tarda
Abstract
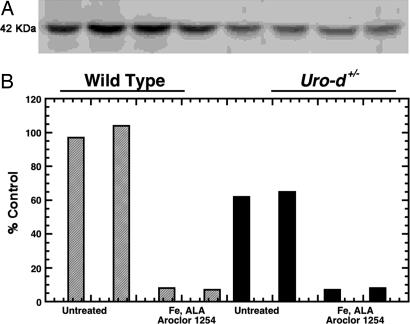
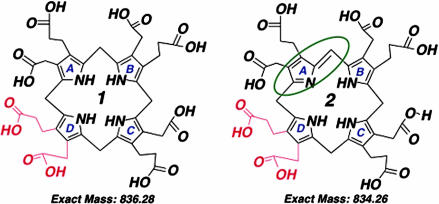
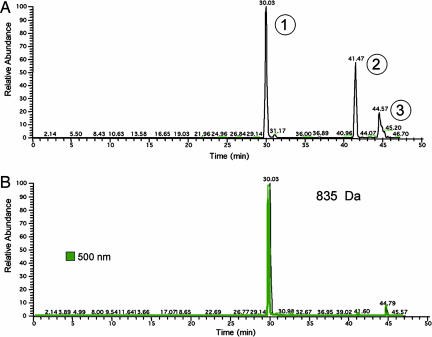
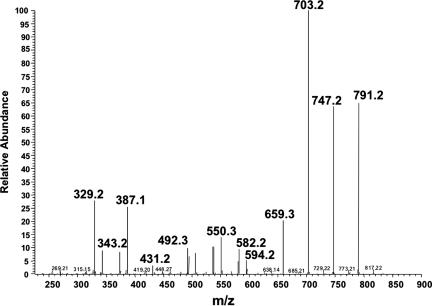
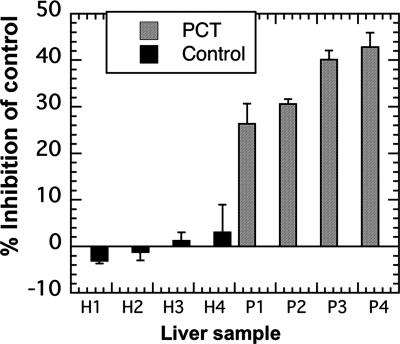
Porphyria cutanea tarda (PCT), the most common form of porphyria in humans, is due to reduced activity of uroporphyrinogen decarboxylase (URO-D) in the liver. Previous studies have demonstrated that protein levels of URO-D do not change when catalytic activity is reduced, suggesting that an inhibitor of URO-D is generated in hepatocytes. Here, we describe the identification and characterization of an inhibitor of URO-D in liver cytosolic extracts from two murine models of PCT: wild-type mice treated with iron, delta-aminolevulinic acid, and polychlorinated biphenyls; and mice with one null allele of Uro-d and two null alleles of the hemochromatosis gene (Uro-d(+/-), Hfe(-/-)) that develop PCT with no treatments. In both models, we identified an inhibitor of recombinant human URO-D (rhURO-D). The inhibitor was characterized by solid-phase extraction, chromatography, UV-visible spectroscopy, and mass spectroscopy and proved to be uroporphomethene, a compound in which one bridge carbon in the uroporphyrinogen macrocycle is oxidized. We synthesized uroporphomethene by photooxidation of enzymatically generated uroporphyrinogen I or III. Both uroporphomethenes inhibited rhURO-D, but the III isomer porphomethene was a more potent inhibitor. Finally, we detected an inhibitor of rhURO-D in cytosolic extracts of liver biopsy samples of patients with PCT. These studies define the mechanism underlying clinical expression of the PCT phenotype, namely oxidation of uroporphyrinogen to uroporphomethene, a competitive inhibitor of URO-D. The oxidation reaction is iron-dependent.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Anderson KE, Sassa S, Bishop DF, Desnick RJ. In: The Metabolic and Molecular Bases of Inherited Disease. Scriver CR, Beaudet AL, Sly WS, Valle D, editors. Vol 2. New York: McGraw–Hill; 2001. pp. 2991–3062.
-
- Bulaj ZJ, Phillips JD, Ajioka RS, Franklin MR, Griffen LM, Guinee DJ, Edwards CQ, Kushner JP. Blood. 2000;95:1565–1571. - PubMed
-
- Felsher BF, Kushner JP. Semin Hematol. 1977;14:243–251. - PubMed
-
- Lundvall O. Acta Med Scand. 1971;189:33–49. - PubMed
-
- Elder GH, Urquhart AJ, De Salamanca RE, Munzo JJ, Bonkovsky HL. Lancet. 1985;ii:229–232. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
