

Structures of the extracellular regions of the group II/III metabotropic glutamate receptors
- PMID: 17360426
- PMCID: PMC1820657
- DOI: 10.1073/pnas.0611577104
Structures of the extracellular regions of the group II/III metabotropic glutamate receptors
Abstract
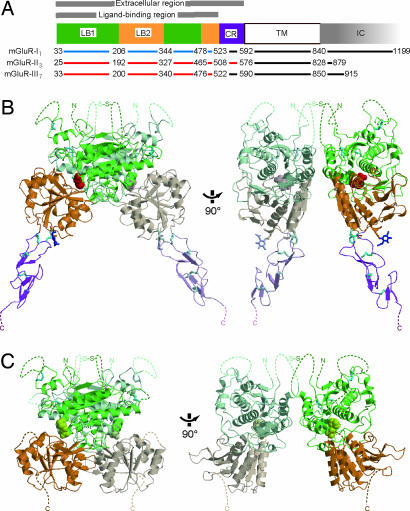
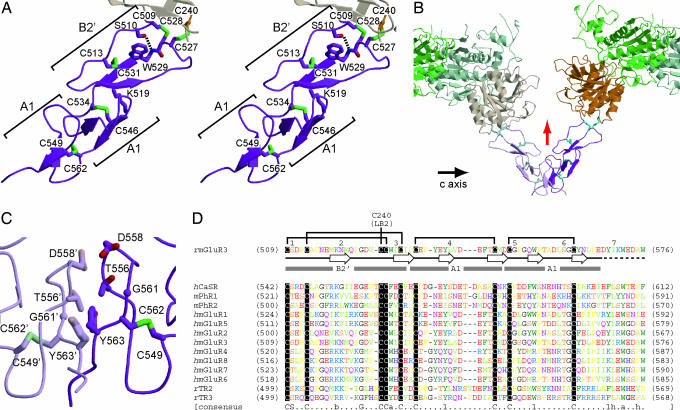
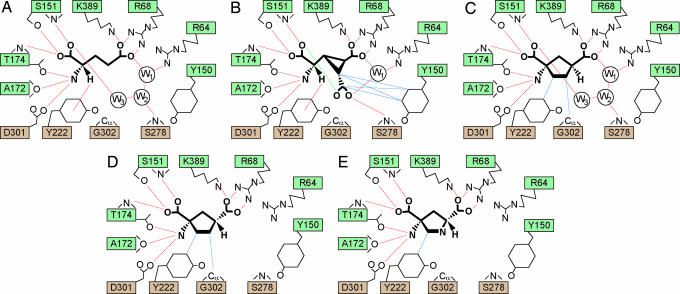
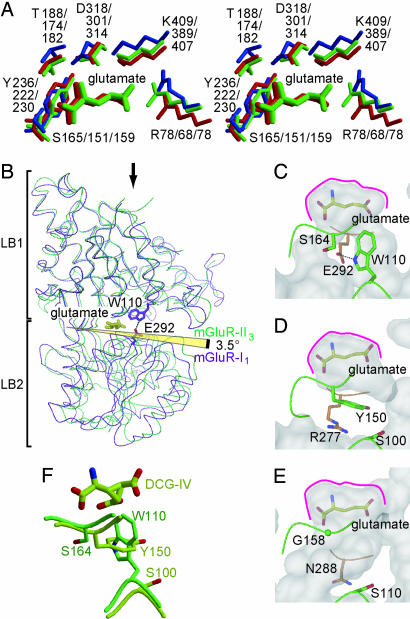
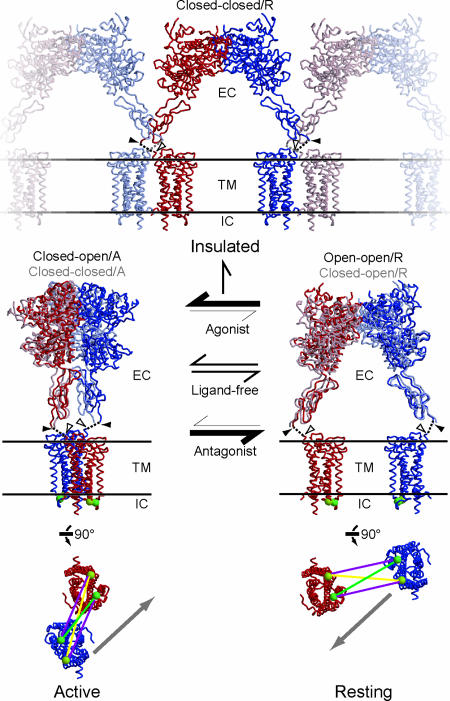
Metabotropic glutamate receptors play major roles in the activation of excitatory synapses in the central nerve system. We determined the crystal structure of the entire extracellular region of the group II receptor and that of the ligand-binding region of the group III receptor. A comparison among groups I, II, and III provides the structural basis that could account for the discrimination of group-specific agonists. Furthermore, the structure of group II includes the cysteine-rich domain, which is tightly linked to the ligand-binding domain by a disulfide bridge, suggesting a potential role in transmitting a ligand-induced conformational change into the downstream transmembrane region. The structure also reveals the lateral interaction between the two cysteine-rich domains, which could stimulate clustering of the dimeric receptors on the cell surface. We propose a general activation mechanism of the dimeric receptor coupled with both ligand-binding and interprotomer rearrangements.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Pin JP, Galvez T, Prézeau L. Pharmacol Ther. 2003;98:325–354. - PubMed
-
- Nakanishi S, Masu M. Annu Rev Biophys Biomol Struct. 1994;23:319–348. - PubMed
-
- Hollmann M, Heinemann S. Annu Rev Neurosci. 1994;17:31–108. - PubMed
-
- Kunishima N, Shimada Y, Tsuji Y, Sato T, Yamamoto M, Kumasaka T, Nakanishi S, Jingami H, Morikawa K. Nature. 2000;407:971–977. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
