

The nuclear function of p53 is required for PUMA-mediated apoptosis induced by DNA damage
- PMID: 17360476
- PMCID: PMC1820707
- DOI: 10.1073/pnas.0700020104
The nuclear function of p53 is required for PUMA-mediated apoptosis induced by DNA damage
Abstract
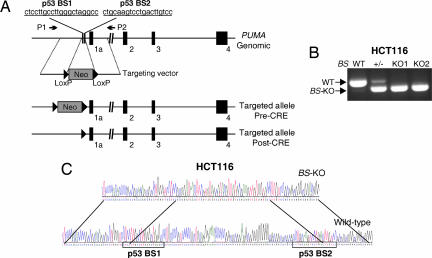
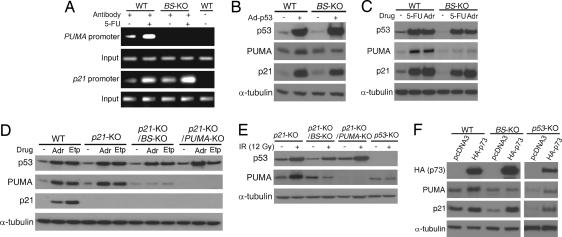
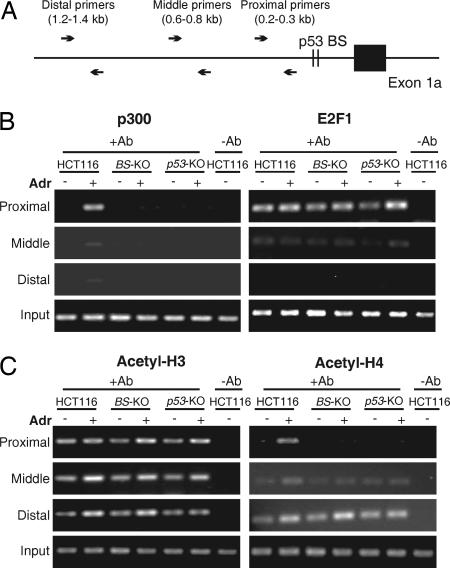
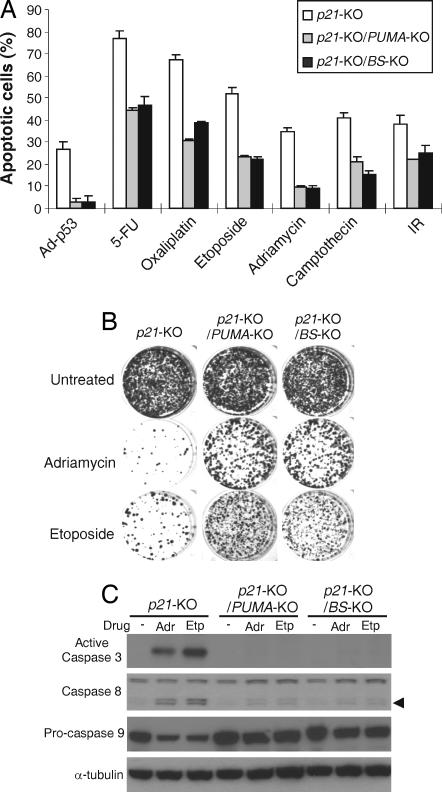
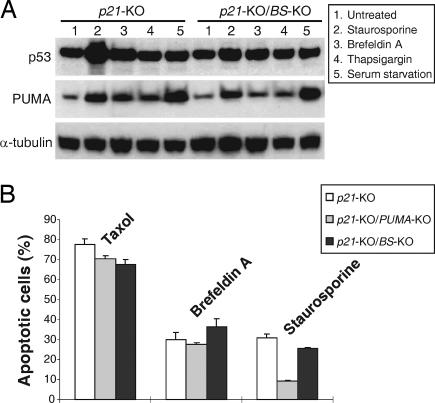
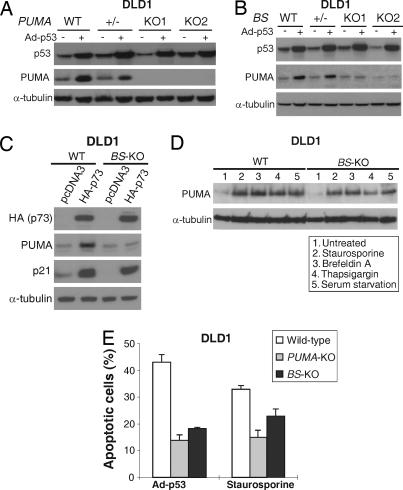
The tumor suppressor p53 can induce apoptosis by activating gene expression in the nucleus, or by directly permeabilizing mitochondria in the cytoplasm. It has been shown that PUMA, a downstream target of p53 and a BH3-only Bcl-2 family member, plays an essential role in apoptosis induced by both nuclear and cytoplasmic p53. To understand how PUMA does so, we used homologous recombination to delete the binding sites of p53 in the promoter of PUMA in human colorectal cancer cells. As a result, the induction of PUMA and apoptosis in response to p53 and DNA-damaging agents were abrogated. Transcription coactivator recruitment and histone modifications in the PUMA promoter were suppressed. However, induction of PUMA and apoptosis in response to non-DNA-damaging stimuli were unaffected. These results indicate that the binding of nuclear p53 to the specific sites within the PUMA promoter is essential for its ability to induce apoptosis and is likely to be required for its tumor suppressive capacity.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
PUMA couples the nuclear and cytoplasmic proapoptotic function of p53.Science. 2005 Sep 9;309(5741):1732-5. doi: 10.1126/science.1114297. Science. 2005. PMID: 16151013
-
Noxa induces apoptosis in oncogene-expressing cells through catch-and-release mechanism operating between Puma and Mcl-1.Biochem Biophys Res Commun. 2011 Oct 7;413(4):643-8. doi: 10.1016/j.bbrc.2011.09.036. Epub 2011 Sep 14. Biochem Biophys Res Commun. 2011. PMID: 21945433
-
Transcription factor Sox4 is required for PUMA-mediated apoptosis induced by histone deacetylase inhibitor, TSA.Biochem Biophys Res Commun. 2013 Aug 23;438(2):445-51. doi: 10.1016/j.bbrc.2013.07.099. Epub 2013 Jul 31. Biochem Biophys Res Commun. 2013. PMID: 23916609
-
PUMA, a potent killer with or without p53.Oncogene. 2008 Dec;27 Suppl 1(Suppl 1):S71-83. doi: 10.1038/onc.2009.45. Oncogene. 2008. PMID: 19641508 Free PMC article. Review.
-
p53 in neuronal apoptosis.Biochem Biophys Res Commun. 2005 Jun 10;331(3):761-77. doi: 10.1016/j.bbrc.2005.03.149. Biochem Biophys Res Commun. 2005. PMID: 15865932 Review.
Cited by
-
Crizotinib induces PUMA-dependent apoptosis in colon cancer cells.Mol Cancer Ther. 2013 May;12(5):777-86. doi: 10.1158/1535-7163.MCT-12-1146. Epub 2013 Feb 20. Mol Cancer Ther. 2013. PMID: 23427294 Free PMC article.
-
In vivo RNA-seq and ChIP-seq analyses show an obligatory role for the C terminus of p53 in conferring tissue-specific radiation sensitivity.Cell Rep. 2023 Mar 28;42(3):112216. doi: 10.1016/j.celrep.2023.112216. Epub 2023 Mar 15. Cell Rep. 2023. PMID: 36924496 Free PMC article.
-
Mutant p53 drives cancer chemotherapy resistance due to loss of function on activating transcription of PUMA.Cell Cycle. 2019 Dec;18(24):3442-3455. doi: 10.1080/15384101.2019.1688951. Epub 2019 Nov 14. Cell Cycle. 2019. PMID: 31726940 Free PMC article.
-
PUMA promotes Bax translocation by both directly interacting with Bax and by competitive binding to Bcl-X L during UV-induced apoptosis.Mol Biol Cell. 2009 Jul;20(13):3077-87. doi: 10.1091/mbc.e08-11-1109. Epub 2009 May 13. Mol Biol Cell. 2009. PMID: 19439449 Free PMC article.
-
PUMA mediates EGFR tyrosine kinase inhibitor-induced apoptosis in head and neck cancer cells.Oncogene. 2009 Jun 18;28(24):2348-57. doi: 10.1038/onc.2009.108. Epub 2009 May 4. Oncogene. 2009. PMID: 19421143 Free PMC article.
References
-
- Kern SE, Kinzler KW, Bruskin A, Jarosz D, Friedman P, Prives C, Vogelstein B. Science. 1991;252:1708–1711. - PubMed
-
- Vogelstein B, Lane D, Levine AJ. Nature. 2000;408:307–310. - PubMed
-
- El-Deiry WS, Kern SE, Pietenpol JA, Kinzler KW, Vogelstein B. Nat Genet. 1992;1:45–49. - PubMed
-
- Tokino T, Thiagalingam S, el-Deiry WS, Waldman T, Kinzler KW, Vogelstein B. Hum Mol Genet. 1994;3:1537–1542. - PubMed
-
- Wei CL, Wu Q, Vega VB, Chiu KP, Ng P, Zhang T, Shahab A, Yong HC, Fu Y, Weng Z, et al. Cell. 2006;124:207–219. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
