

Regulation of Alzheimer's disease amyloid-beta formation by casein kinase I
- PMID: 17360493
- PMCID: PMC1820725
- DOI: 10.1073/pnas.0611236104
Regulation of Alzheimer's disease amyloid-beta formation by casein kinase I
Abstract
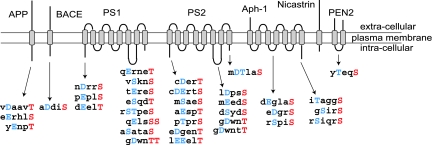
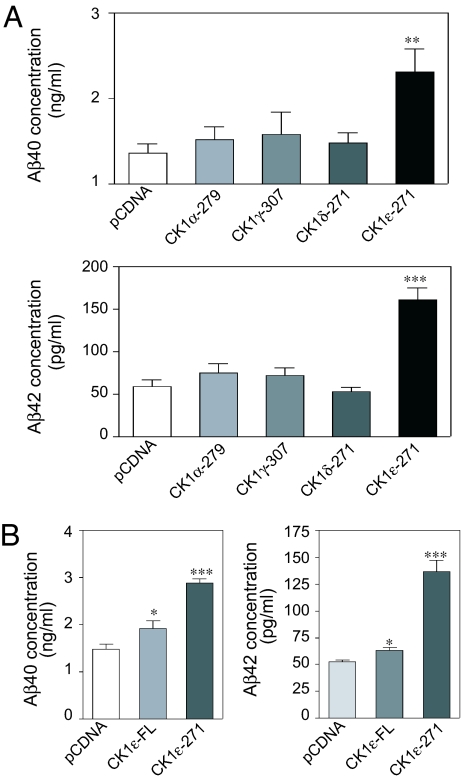
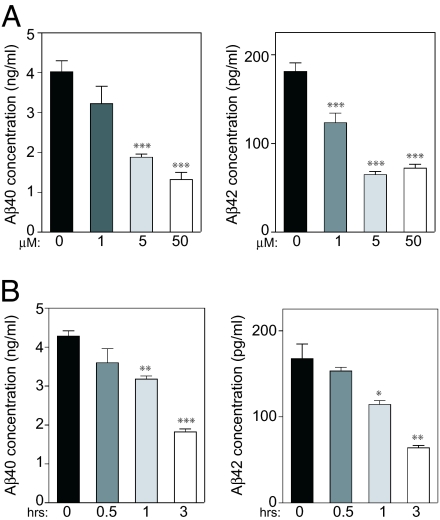
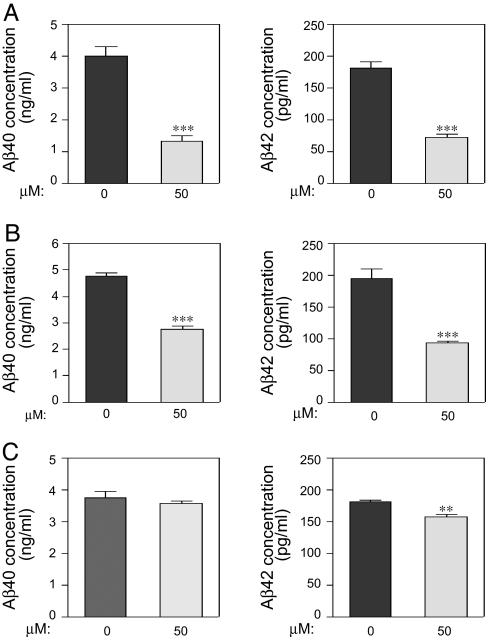
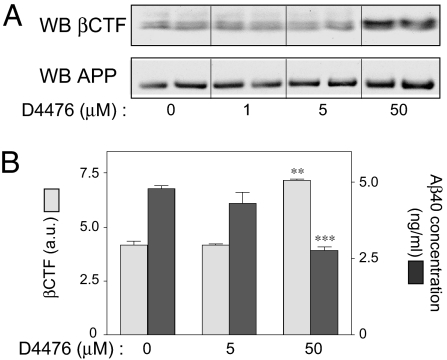
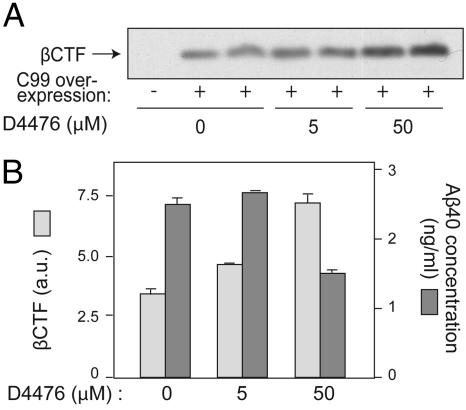
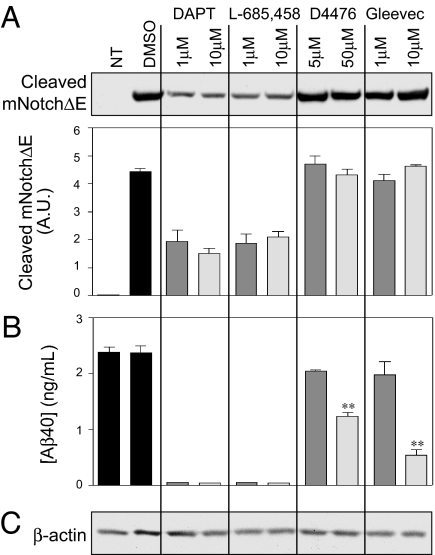
Alzheimer's disease (AD) is associated with accumulation of the neurotoxic peptide amyloid-beta (Abeta), which is produced by sequential cleavage of amyloid precursor protein (APP) by the aspartyl protease beta-secretase and the presenilin-dependent protease gamma-secretase. An increase of casein kinase 1 (CK1) expression has been described in the human AD brain. We show, by using in silico analysis, that APP, beta-secretase, and gamma-secretase subunits contain, in their intracellular regions, multiple CK1 consensus phosphorylation sites, many of which are conserved among human, rat, and mouse species. Overexpression of constitutively active CK1epsilon, one of the CK1 isoforms expressed in brain, leads to an increase in Abeta peptide production. Conversely, three structurally dissimilar CK1-specific inhibitors significantly reduced endogenous Abeta peptide production. By using mammalian cells expressing the beta C-terminal fragment of APP, it was possible to demonstrate that CK1 inhibitors act at the level of gamma-secretase cleavage. Importantly, Notch cleavage was not affected. Our results indicate that CK1 represents a therapeutic target for prevention of Abeta formation in AD.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Yasojima K, Kuret J, DeMaggio AJ, McGeer E, McGeer PL. Brain Res. 2000;865:116–120. - PubMed
-
- Schwab C, DeMaggio AJ, Ghoshal N, Binder LI, Kuret J, McGeer PL. Neurobiol Aging. 2000;21:503–510. - PubMed
-
- Phiel CJ, Wilson CA, Lee VM, Klein PS. Nature. 2003;423:435–439. - PubMed
-
- Meijer L, Flajolet M, Greengard P. Trends Pharmacol Sci. 2004;25:471–480. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
