

Mitotic retention of gene expression patterns by the cell fate-determining transcription factor Runx2
- PMID: 17360627
- PMCID: PMC1805558
- DOI: 10.1073/pnas.0611419104
Mitotic retention of gene expression patterns by the cell fate-determining transcription factor Runx2
Abstract
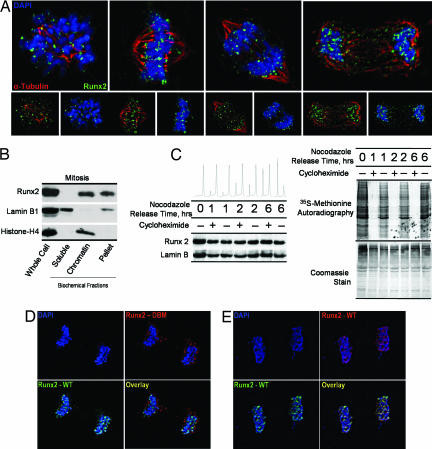
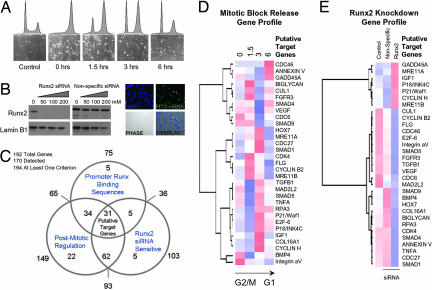
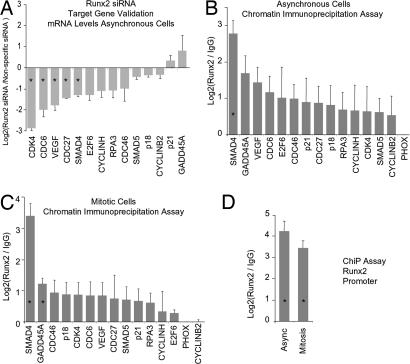
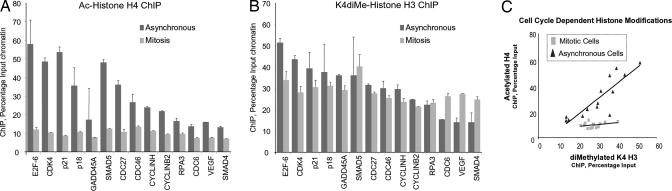
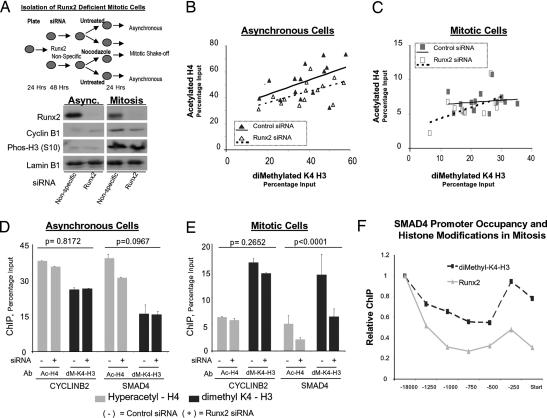
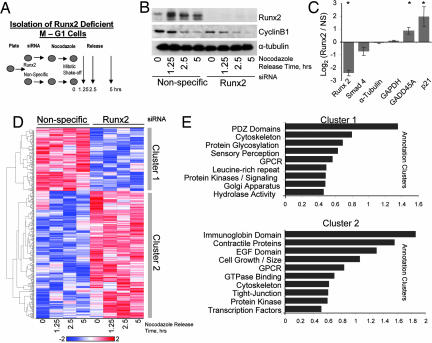
During cell division, cessation of transcription is coupled with mitotic chromosome condensation. A fundamental biological question is how gene expression patterns are retained during mitosis to ensure the phenotype of progeny cells. We suggest that cell fate-determining transcription factors provide an epigenetic mechanism for the retention of gene expression patterns during cell division. Runx proteins are lineage-specific transcription factors that are essential for hematopoietic, neuronal, gastrointestinal, and osteogenic cell fates. Here we show that Runx2 protein is stable during cell division and remains associated with chromosomes during mitosis through sequence-specific DNA binding. Using siRNA-mediated silencing, mitotic cell synchronization, and expression profiling, we identify Runx2-regulated genes that are modulated postmitotically. Novel target genes involved in cell growth and differentiation were validated by chromatin immunoprecipitation. Importantly, we find that during mitosis, when transcription is shut down, Runx2 selectively occupies target gene promoters, and Runx2 deficiency alters mitotic histone modifications. We conclude that Runx proteins have an active role in retaining phenotype during cell division to support lineage-specific control of gene expression in progeny cells.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Mitotic occupancy and lineage-specific transcriptional control of rRNA genes by Runx2.Nature. 2007 Jan 25;445(7126):442-6. doi: 10.1038/nature05473. Nature. 2007. PMID: 17251981
-
Mitotic Inheritance of mRNA Facilitates Translational Activation of the Osteogenic-Lineage Commitment Factor Runx2 in Progeny of Osteoblastic Cells.J Cell Physiol. 2016 May;231(5):1001-14. doi: 10.1002/jcp.25188. Epub 2015 Sep 18. J Cell Physiol. 2016. PMID: 26381402 Free PMC article.
-
Live cell imaging of the cancer-related transcription factor RUNX2 during mitotic progression.J Cell Physiol. 2011 May;226(5):1383-9. doi: 10.1002/jcp.22465. J Cell Physiol. 2011. PMID: 20945391 Free PMC article.
-
Transcription factor retention on mitotic chromosomes: regulatory mechanisms and impact on cell fate decisions.FEBS Lett. 2018 Mar;592(6):878-887. doi: 10.1002/1873-3468.12828. Epub 2017 Sep 12. FEBS Lett. 2018. PMID: 28862742 Review.
-
Mitotic Gene Bookmarking: An Epigenetic Mechanism for Coordination of Lineage Commitment, Cell Identity and Cell Growth.Adv Exp Med Biol. 2017;962:95-102. doi: 10.1007/978-981-10-3233-2_7. Adv Exp Med Biol. 2017. PMID: 28299653 Free PMC article. Review.
Cited by
-
Comprehensive expression map of transcription regulators in the adult zebrafish telencephalon reveals distinct neurogenic niches.J Comp Neurol. 2015 Jun 1;523(8):1202-21. doi: 10.1002/cne.23733. Epub 2015 Feb 19. J Comp Neurol. 2015. PMID: 25556858 Free PMC article.
-
Inhibition of the RUNX1-CBFβ transcription factor complex compromises mammary epithelial cell identity: a phenotype potentially stabilized by mitotic gene bookmarking.Oncotarget. 2020 Jun 30;11(26):2512-2530. doi: 10.18632/oncotarget.27637. eCollection 2020 Jun 30. Oncotarget. 2020. PMID: 32655837 Free PMC article.
-
The abbreviated pluripotent cell cycle.J Cell Physiol. 2013 Jan;228(1):9-20. doi: 10.1002/jcp.24104. J Cell Physiol. 2013. PMID: 22552993 Free PMC article. Review.
-
Similarities between the Epstein-Barr Virus (EBV) Nuclear Protein EBNA1 and the Pioneer Transcription Factor FoxA: Is EBNA1 a "Bookmarking" Oncoprotein that Alters the Host Cell Epigenotype?Pathogens. 2012 Sep 17;1(1):37-51. doi: 10.3390/pathogens1010037. Pathogens. 2012. PMID: 25436603 Free PMC article. Review.
-
The osteogenic transcription factor runx2 controls genes involved in sterol/steroid metabolism, including CYP11A1 in osteoblasts.Mol Endocrinol. 2009 Jun;23(6):849-61. doi: 10.1210/me.2008-0270. Epub 2009 Apr 2. Mol Endocrinol. 2009. PMID: 19342447 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
