

NMDA receptor subunits have differential roles in mediating excitotoxic neuronal death both in vitro and in vivo
- PMID: 17360906
- PMCID: PMC6672582
- DOI: 10.1523/JNEUROSCI.0116-07.2007
NMDA receptor subunits have differential roles in mediating excitotoxic neuronal death both in vitro and in vivo
Abstract
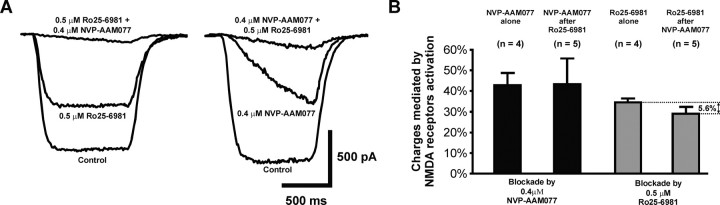
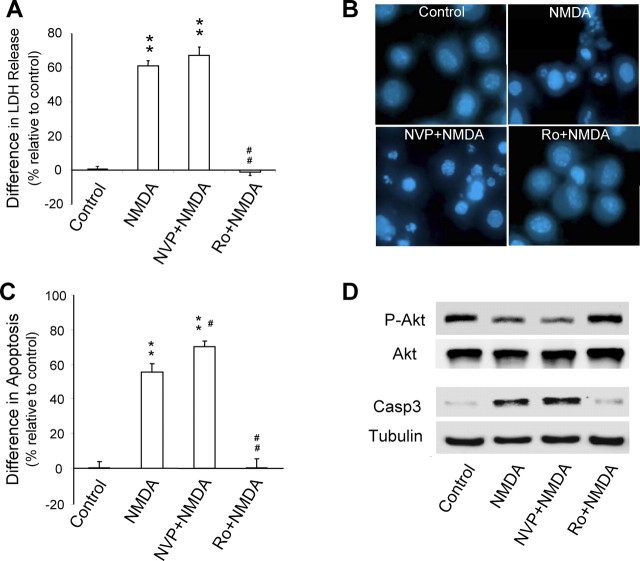
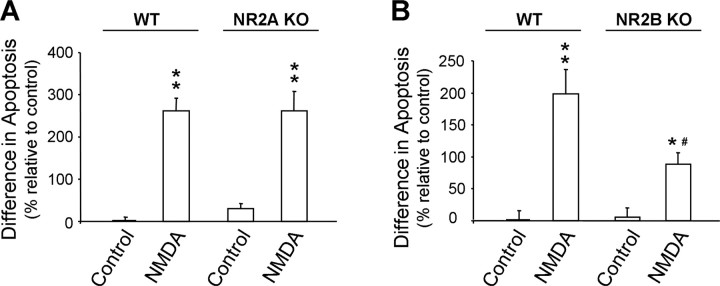
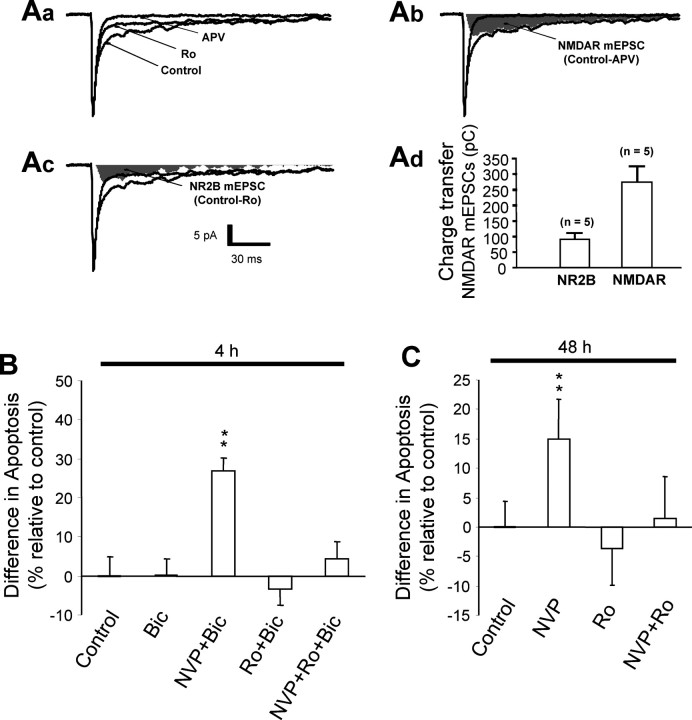
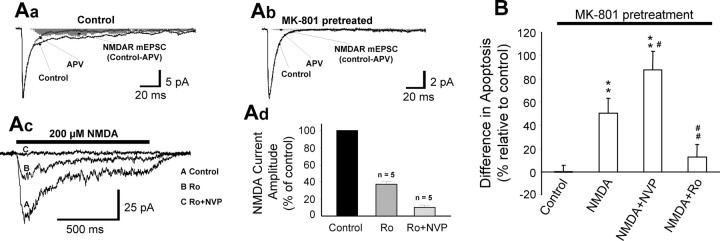
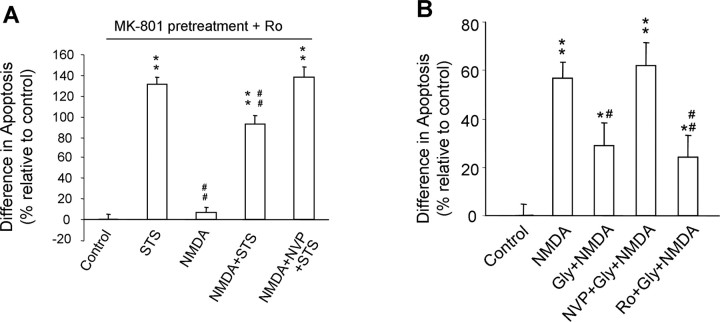
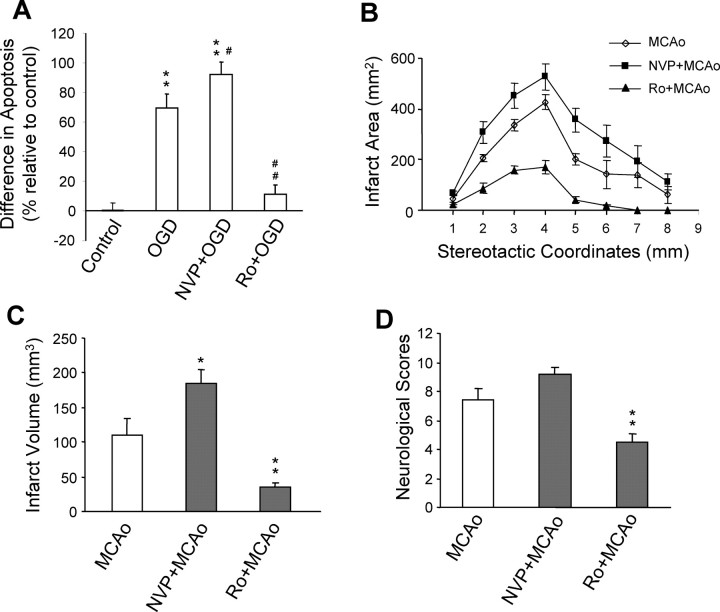
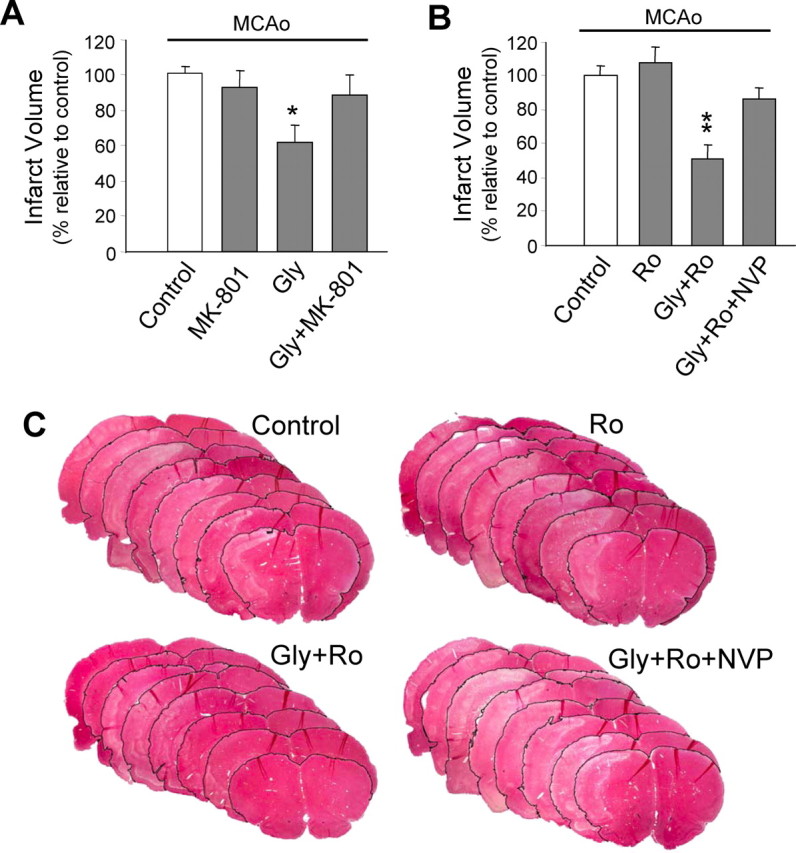
Well-documented experimental evidence from both in vitro and in vivo models of stroke strongly supports the critical involvement of NMDA receptor-mediated excitotoxicity in neuronal damage after stroke. Despite this, the results of clinical trials testing NMDA receptor antagonists as neuroprotectants after stroke and brain trauma have been discouraging. Here, we report that in mature cortical cultures, activation of either synaptic or extrasynaptic NR2B-containing NMDA receptors results in excitotoxicity, increasing neuronal apoptosis. In contrast, activation of either synaptic or extrasynaptic NR2A-containing NMDA receptors promotes neuronal survival and exerts a neuroprotective action against both NMDA receptor-mediated and non-NMDA receptor-mediated neuronal damage. A similar opposing action of NR2B and NR2A in mediating cell death and cell survival was also observed in an in vivo rat model of focal ischemic stroke. Moreover, we found that blocking NR2B-mediated cell death was effective in reducing infarct volume only when the receptor antagonist was given before the onset of stroke and not 4.5 h after stroke. In great contrast, activation of NR2A-mediated cell survival signaling with administration of either glycine alone or in the presence of NR2B antagonist significantly attenuated ischemic brain damage even when delivered 4.5 h after stroke onset. Together, the present work provides a molecular basis for the dual roles of NMDA receptors in promoting neuronal survival and mediating neuronal damage and suggests that selective enhancement of NR2A-containing NMDA receptor activation with glycine may constitute a promising therapy for stroke.
Figures
Similar articles
-
In developing hippocampal neurons, NR2B-containing N-methyl-D-aspartate receptors (NMDARs) can mediate signaling to neuronal survival and synaptic potentiation, as well as neuronal death.Neuroscience. 2009 Jan 12;158(1):334-43. doi: 10.1016/j.neuroscience.2008.01.080. Epub 2008 Mar 4. Neuroscience. 2009. PMID: 18378405 Free PMC article.
-
Excitotoxicity in vitro by NR2A- and NR2B-containing NMDA receptors.Neuropharmacology. 2007 Jul;53(1):10-7. doi: 10.1016/j.neuropharm.2007.04.015. Epub 2007 May 8. Neuropharmacology. 2007. PMID: 17570444
-
Functional NMDA receptor subtype 2B is expressed in astrocytes after ischemia in vivo and anoxia in vitro.J Neurosci. 2003 Apr 15;23(8):3364-72. doi: 10.1523/JNEUROSCI.23-08-03364.2003. J Neurosci. 2003. PMID: 12716944 Free PMC article.
-
The rise and fall of NMDA antagonists for ischemic stroke.Curr Mol Med. 2004 Mar;4(2):131-6. doi: 10.2174/1566524043479248. Curr Mol Med. 2004. PMID: 15032709 Review.
-
NMDA/NR2B selective antagonists in the treatment of ischemic brain injury.Curr Drug Targets CNS Neurol Disord. 2005 Apr;4(2):143-51. doi: 10.2174/1568007053544183. Curr Drug Targets CNS Neurol Disord. 2005. PMID: 15857299 Review.
Cited by
-
Electroacupuncture Attenuates Cerebral Ischemia and Reperfusion Injury in Middle Cerebral Artery Occlusion of Rat via Modulation of Apoptosis, Inflammation, Oxidative Stress, and Excitotoxicity.Evid Based Complement Alternat Med. 2016;2016:9438650. doi: 10.1155/2016/9438650. Epub 2016 Mar 31. Evid Based Complement Alternat Med. 2016. PMID: 27123035 Free PMC article.
-
Differential roles of GluN2A- and GluN2B-containing NMDA receptors in neuronal survival and death.Int J Physiol Pathophysiol Pharmacol. 2012;4(4):211-8. Epub 2012 Dec 26. Int J Physiol Pathophysiol Pharmacol. 2012. PMID: 23320134 Free PMC article.
-
The effects of sigma (σ1) receptor-selective ligands on muscarinic receptor antagonist-induced cognitive deficits in mice.Br J Pharmacol. 2015 May;172(10):2519-31. doi: 10.1111/bph.13076. Epub 2015 Apr 10. Br J Pharmacol. 2015. PMID: 25573298 Free PMC article.
-
Early mechanisms of pathobiology are revealed by transcriptional temporal dynamics in hippocampal CA1 neurons of prion infected mice.PLoS Pathog. 2012;8(11):e1003002. doi: 10.1371/journal.ppat.1003002. Epub 2012 Nov 8. PLoS Pathog. 2012. PMID: 23144617 Free PMC article.
-
Neuroprotective effects of MK-801 against cerebral ischemia reperfusion.Heliyon. 2024 Jun 28;10(13):e33821. doi: 10.1016/j.heliyon.2024.e33821. eCollection 2024 Jul 15. Heliyon. 2024. PMID: 39040387 Free PMC article.
References
-
- Aarts M, Liu Y, Liu L, Besshoh S, Arundine M, Gurd JW, Wang YT, Salter MW, Tymianski M. Treatment of ischemic brain damage by perturbing NMDA receptor-PSD-95 protein interactions. Science. 2002;298:846–850. - PubMed
-
- Aarts M, Iihara K, Wei WL, Xiong ZG, Arundine M, Cerwinski W, MacDonald JF, Tymianski M. A key role for TRPM7 channels in anoxic neuronal death. Cell. 2003;115:863–877. - PubMed
-
- Bederson JB, Pitts LH, Tsuji M, Nishimura MC, Davis RL, Bartkowski H. Rat middle cerebral artery occlusion: evaluation of the model and development of a neurologic examination. Stroke. 1986;17:472–476. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources