

Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway
- PMID: 17360908
- PMCID: PMC6672572
- DOI: 10.1523/JNEUROSCI.4970-06.2007
Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway
Abstract
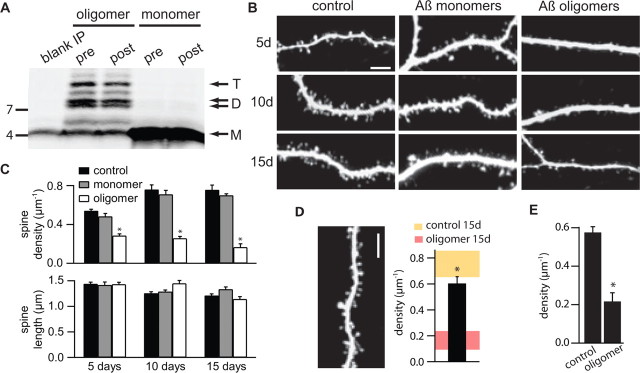
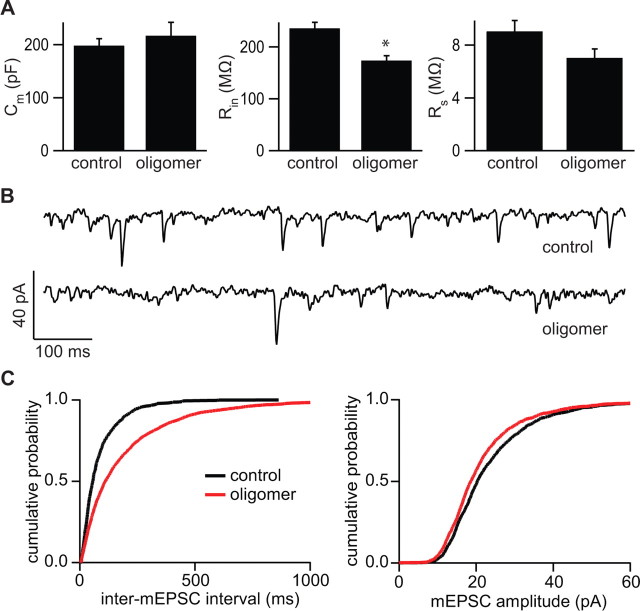
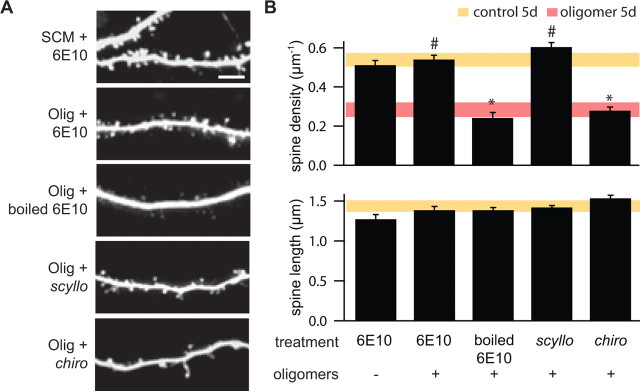
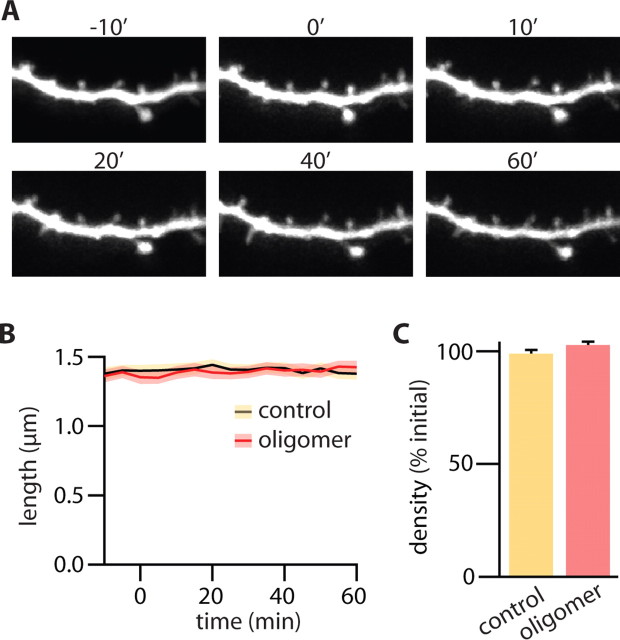
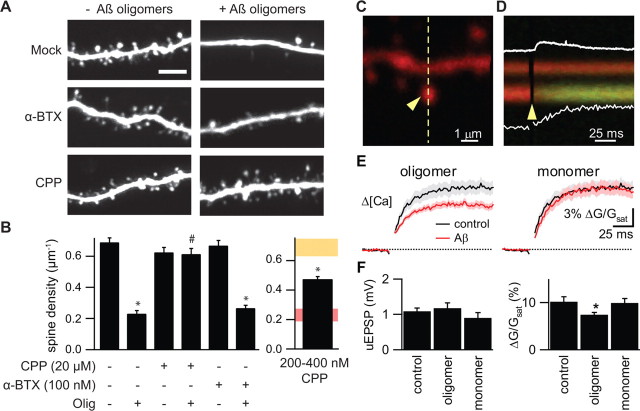
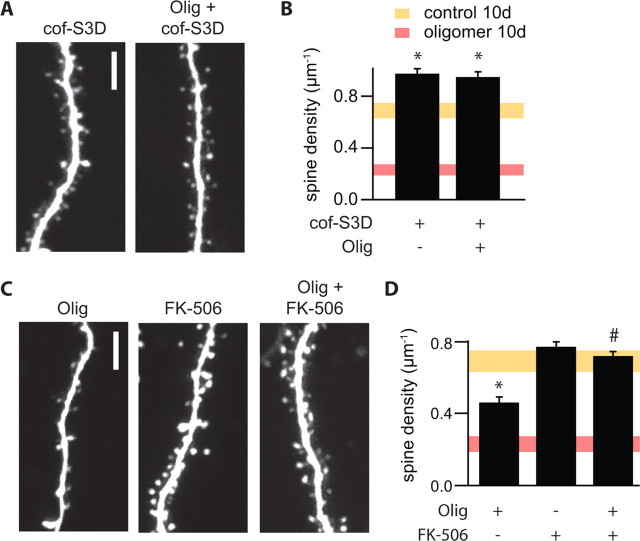
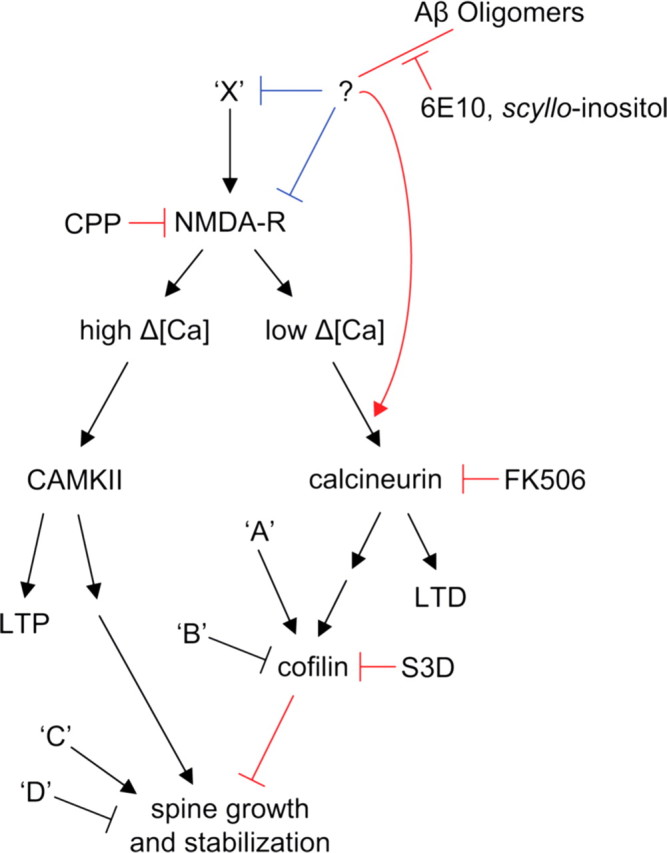
Alzheimer's disease (AD) is characterized by decreased synapse density in hippocampus and neocortex, and synapse loss is the strongest anatomical correlate of the degree of clinical impairment. Although considerable evidence supports a causal role for the amyloid-beta protein (Abeta) in AD, a direct link between a specific form of Abeta and synapse loss has not been established. We demonstrate that physiological concentrations of naturally secreted Abeta dimers and trimers, but not monomers, induce progressive loss of hippocampal synapses. Pyramidal neurons in rat organotypic slices had markedly decreased density of dendritic spines and numbers of electrophysiologically active synapses after exposure to picomolar levels of soluble oligomers. Spine loss was reversible and was prevented by Abeta-specific antibodies or a small-molecule modulator of Abeta aggregation. Mechanistically, Abeta-mediated spine loss required activity of NMDA-type glutamate receptors (NMDARs) and occurred through a pathway involving cofilin and calcineurin. Furthermore, NMDAR-mediated calcium influx into active spines was reduced by Abeta oligomers. Partial blockade of NMDARs by pharmacological antagonists was sufficient to trigger spine loss. We conclude that soluble, low-n oligomers of human Abeta trigger synapse loss that can be reversed by therapeutic agents. Our approach provides a quantitative cellular model for elucidating the molecular basis of Abeta-induced neuronal dysfunction.
Figures
References
-
- Bloodgood BL, Sabatini BL. Nonlinear regulation of unitary synaptic signals by CaV(2.3) voltage-sensitive calcium channels located in dendritic spines. Neuron. 2007;53:249–260. - PubMed
-
- Carter AG, Sabatini BL. State-dependent calcium signaling in dendritic spines of striatal medium spiny neurons. Neuron. 2004;44:483–493. - PubMed
-
- Chen QS, Wei WZ, Shimahara T, Xie CW. Alzheimer amyloid beta-peptide inhibits the late phase of long-term potentiation through calcineurin-dependent mechanisms in the hippocampal dentate gyrus. Neurobiol Learn Mem. 2002;77:354–371. - PubMed
-
- Cleary JP, Walsh DM, Hofmeister JJ, Shankar GM, Kuskowski MA, Selkoe DJ, Ashe KH. Natural oligomers of the amyloid-beta protein specifically disrupt cognitive function. Nat Neurosci. 2005;8:79–84. - PubMed
-
- Cummings JA, Mulkey RM, Nicoll RA, Malenka RC. Ca2+ signaling requirements for long-term depression in the hippocampus. Neuron. 1996;16:825–833. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases