

Genomic deletion within GLDC is a major cause of non-ketotic hyperglycinaemia
- PMID: 17361008
- PMCID: PMC2598024
- DOI: 10.1136/jmg.2006.043448
Genomic deletion within GLDC is a major cause of non-ketotic hyperglycinaemia
Abstract
Background: Non-ketotic hyperglycinaemia (NKH) is an inborn error of metabolism characterised by accumulation of glycine in body fluids and various neurological symptoms. NKH is caused by deficiency of the glycine cleavage multienzyme system with three specific components encoded by GLDC, AMT and GCSH. Most patients are deficient of the enzymatic activity of glycine decarboxylase, which is encoded by GLDC. Our recent study has suggested that there are a considerable number of GLDC mutations which are not identified by the standard exon-sequencing method.
Methods: A screening system for GLDC deletions by multiplex ligation-dependent probe amplification (MLPA) has been developed. Two distinct cohorts of patients with typical NKH were screened by this
Method: the first cohort consisted of 45 families with no identified AMT or GCSH mutations, and the second cohort was comprised of 20 patients from the UK who were not prescreened for AMT mutations.
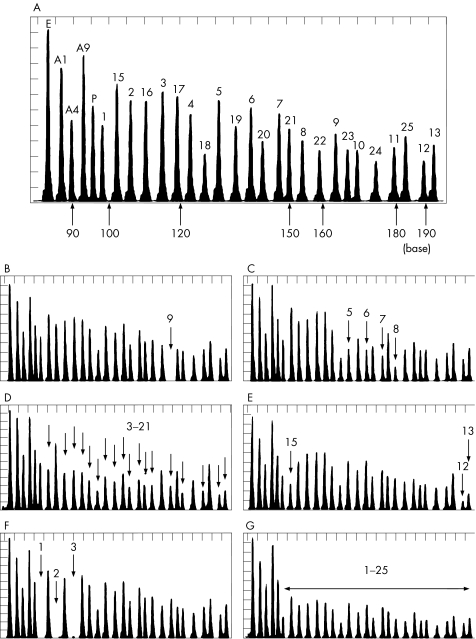
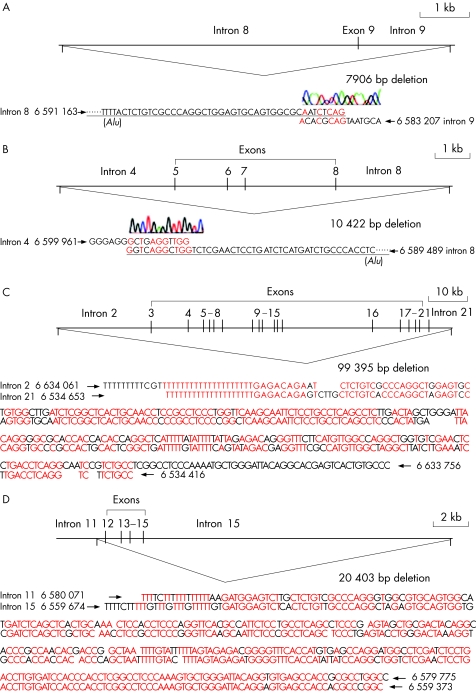
Results: GLDC deletions were identified in 16 of 90 alleles (18%) in the first cohort and in 9 of 40 alleles (22.5%) in the second cohort. 14 different types of deletions of various lengths were identified, including one allele where all 25 exons were missing. Flanking sequences of interstitial deletions in five patients were determined, and Alu-mediated recombination was identified in three of five patients.
Conclusions: GLDC deletions are a significant cause of NKH, and the MLPA analysis is a valuable first-line screening for NKH genetic testing.
Conflict of interest statement
Competing interests: None declared.

References
-
- Hamosh A, Johnston M V. Nonketotic hyperglycinemia. In: Scriver CR, Beaudet AL, Sly WS, et al eds. The metabolic and molecular bases of inherited disease. 8th edn, Vol 2. New York: McGraw‐Hill, 20012065–2078.
-
- Kure S, Tada K, Narisawa K. Nonketotic hyperglycinemia: biochemical, molecular, and neurological aspects. J Hum Genet 19974213–22. - PubMed
-
- Tada K, Kure S. Nonketotic hyperglycinemia: pathophysiological studies. Proc Japan Acad 200581(Ser B)411–417.
-
- Hoover‐Fong J E, Shah S, Van Hove J L, Applegarth D, Toone J, Hamosh A. Natural history of nonketotic hyperglycinemia in 65 patients. Neurology 2004631847–1853. - PubMed
-
- Dinopoulos A, Kure S, Chuck G, Sato K, Gilbert D L, Matsubara Y, Degrauw T. Glycine decarboxylase mutations: a distinctive phenotype of nonketotic hyperglycinemia in adults. Neurology 2005641255–1257. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
LinkOut - more resources
Full Text Sources