

The molecular neurobiology of depression
- PMID: 17362799
- PMCID: PMC2136411
- DOI: 10.1016/j.psc.2006.12.005
The molecular neurobiology of depression
Abstract
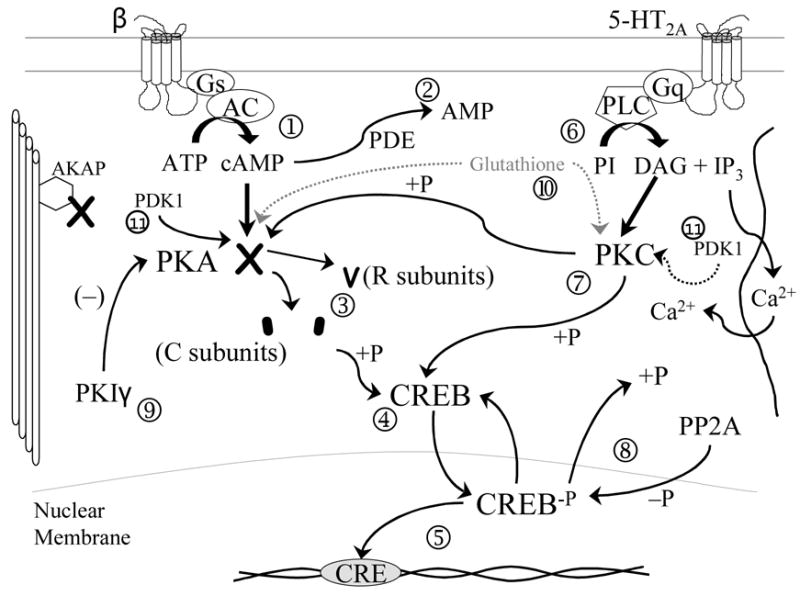
Depression is a condition with a complex biologic pattern in etiology. Environmental stressors modulate subsequent vulnerability to depression. In particular, early adversity seems to induce heightened reactivity to stress through several possible mechanisms, both biologic and psychologic. This increased reactivity results in an enhancement of biologic stress-response mechanisms, especially the HPA axis. Regulators of this system, particularly signal transduction pathways involving PKA and PKC, may be important in the regulation of key genes in this system including genes for GR, BDNF, and trk-b. This system potentially is vulnerable to ROS and therefore, indirectly, to the effects of cytokines. Finally, some of these effects may be controlled by chemical modification of DNA, specifically, methylation of promoters or other gene regions. This modification is a mechanism by which long-term biologic change can be induced by environmental stressors. The brain is homeostatic, and it is possible that alterations at multiple points in this system may induce dysregulation and, as a result, vulnerability to stress. Therefore, a person may be vulnerable to depression, which may be a final common "pathway" for this family of conditions. Individuals may very considerably with regard to the locus of the problem, however. For example, functional variants in a set of genes might predispose some people to depression; others may have epigenetic imprinting; and yet different causes may be at work in others. Although this mix is complicated, it can be unraveled. Doing so could lead to the development of novel interventions that could target specific points of vulnerability, allowing an improved matching of patient to treatment based on differential abnormalities at the cellular level.
Figures
References
-
- Gillespie CF, Nemeroff CB. Hypercortisolemia and depression. Psychosom Med. 2005;67 (Suppl 1):S26–S28. - PubMed
-
- Hayley S, Poulter MO, Merali Z, Anisman H. The pathogenesis of clinical depression: Stressor- and cytokine-induced alterations of neuroplasticity. Neuroscience. 2005;135(3):659–678. - PubMed
-
- Kendler KS, Davis CG, Kessler RC. The familial aggregation of common psychiatric and substance use disorders in the National Comorbidity Survey: a family history study. Br J Psychiatry. 1997;170:541–548. - PubMed
-
- Kendler KS, Kessler RC, Walters EE, MacLean C, Neale MC, Heath AC, et al. Stressful life events, genetic liability, and onset of an episode of major depression in women. Am J Psychiatry. 1995 Jun;153:833–842. - PubMed
-
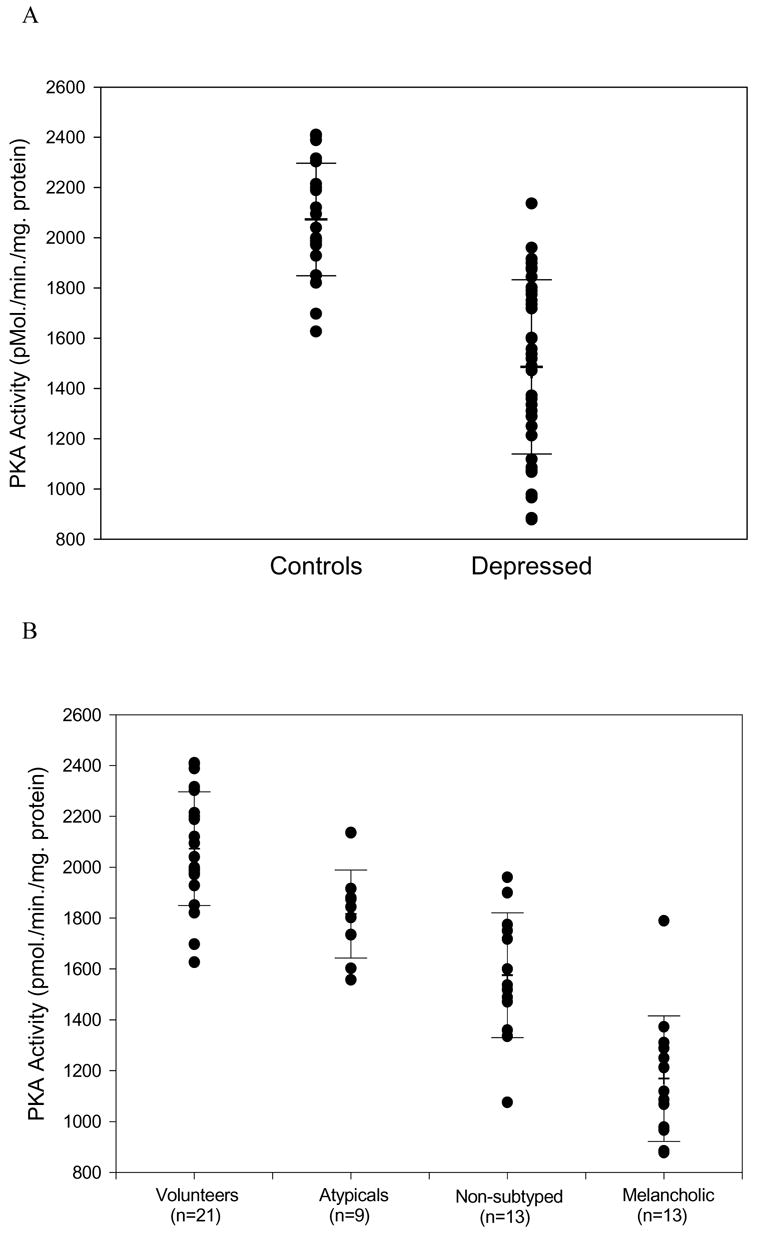
- Shelton RC, Manier DH, Peterson CS, Ellis TC, Sulser F. Cyclic AMP-dependent protein kinase in subtypes of major depression and normal volunteers. Int J Neuropsychopharmcol. 1999;2(3):187–192. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
