

Targeting autophagy augments the anticancer activity of the histone deacetylase inhibitor SAHA to overcome Bcr-Abl-mediated drug resistance
- PMID: 17363733
- PMCID: PMC1896119
- DOI: 10.1182/blood-2006-10-050260
Targeting autophagy augments the anticancer activity of the histone deacetylase inhibitor SAHA to overcome Bcr-Abl-mediated drug resistance
Abstract
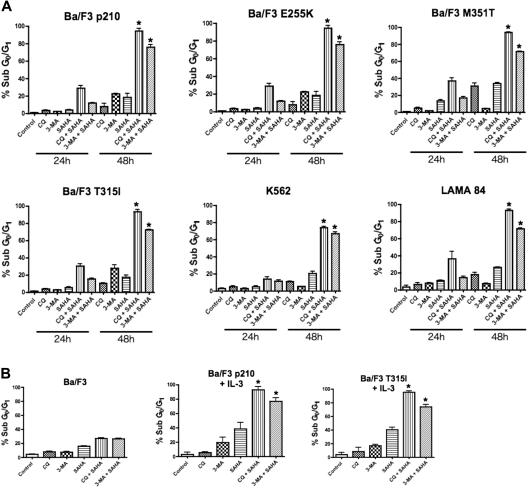
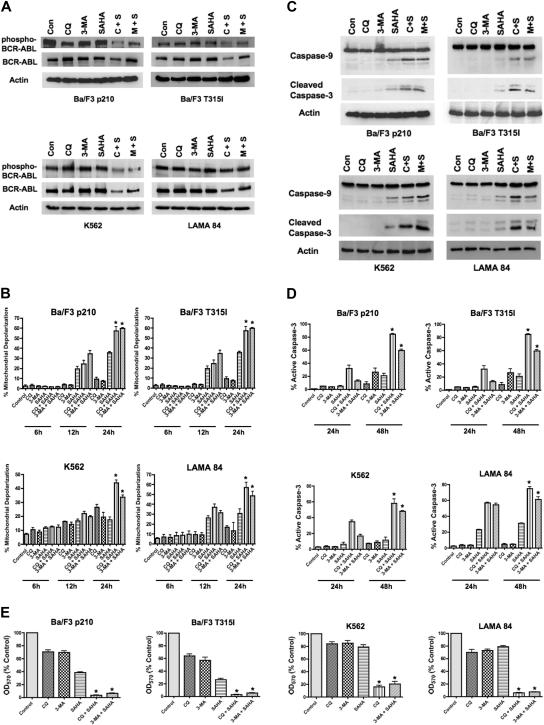
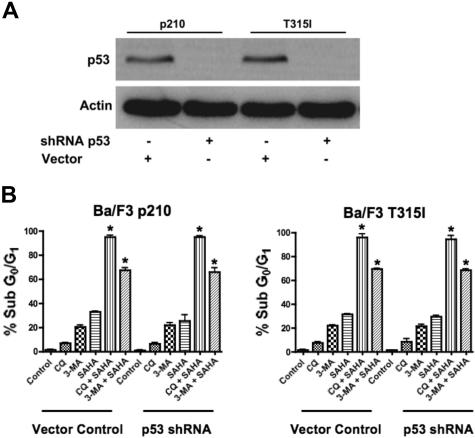
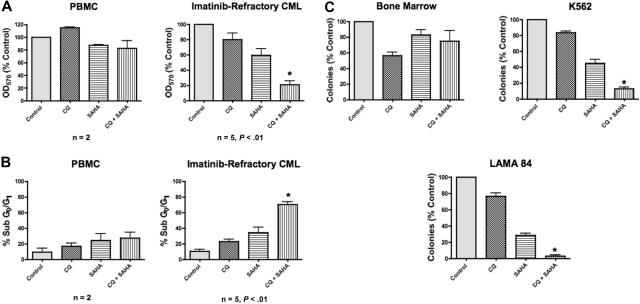
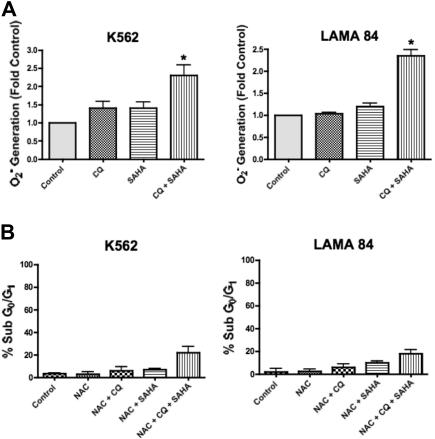
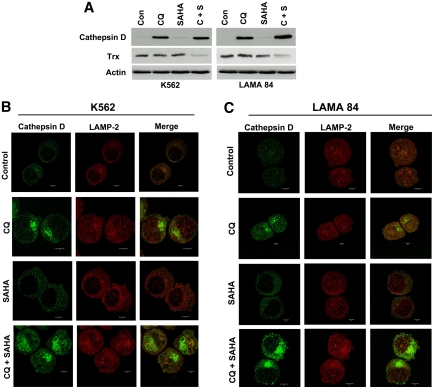
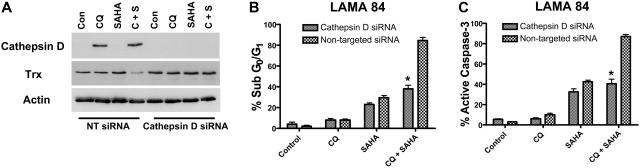
Novel therapeutic strategies are needed to address the emerging problem of imatinib resistance. The histone deacetylase (HDAC) inhibitor suberoylanilide hydroxamic acid (SAHA) is being evaluated for imatinib-resistant chronic myelogenous leukemia (CML) and has multiple cellular effects, including the induction of autophagy and apoptosis. Considering that autophagy may promote cancer cell survival, we hypothesized that disrupting autophagy would augment the anticancer activity of SAHA. Here we report that drugs that disrupt the autophagy pathway dramatically augment the antineoplastic effects of SAHA in CML cell lines and primary CML cells expressing wild-type and imatinib-resistant mutant forms of Bcr-Abl, including T315I. This regimen has selectivity for malignant cells and its efficacy was not diminished by impairing p53 function, another contributing factor in imatinib resistance. Disrupting autophagy by chloroquine treatment enhances SAHA-induced superoxide generation, triggers relocalization and marked increases in the lysosomal protease cathepsin D, and reduces the expression of the cathepsin-D substrate thioredoxin. Finally, knockdown of cathepsin D diminishes the potency of this combination, demonstrating its role as a mediator of this therapeutic response. Our data suggest that, when combined with HDAC inhibitors, agents that disrupt autophagy are a promising new strategy to treat imatinib-refractory patients who fail conventional therapy.
Figures
References
-
- Shah NP, Sawyers CL. Recent success with the tyrosine kinase inhibitor STI-571–lessons for targeted therapy of cancer. Curr Opin Investig Drugs. 2001;2:422–423. - PubMed
-
- Shah NP, Nicoll JM, Nagar B, et al. Multiple BCR-ABL kinase domain mutations confer polyclonal resistance to the tyrosine kinase inhibitor imatinib (STI571) in chronic phase and blast crisis chronic myeloid leukemia. Cancer Cell. 2002;2:117–125. - PubMed
-
- Shah NP, Sawyers CL. Mechanisms of resistance to STI571 in Philadelphia chromosome-associated leukemias. Oncogene. 2003;22:7389–7395. - PubMed
-
- O'Hare T, Corbin AS, Druker BJ. Targeted CML therapy: controlling drug resistance, seeking cure. Curr Opin Genet Dev. 2006;16:92–99. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
