

Global discriminative learning for higher-accuracy computational gene prediction
- PMID: 17367206
- PMCID: PMC1828702
- DOI: 10.1371/journal.pcbi.0030054
Global discriminative learning for higher-accuracy computational gene prediction
Abstract
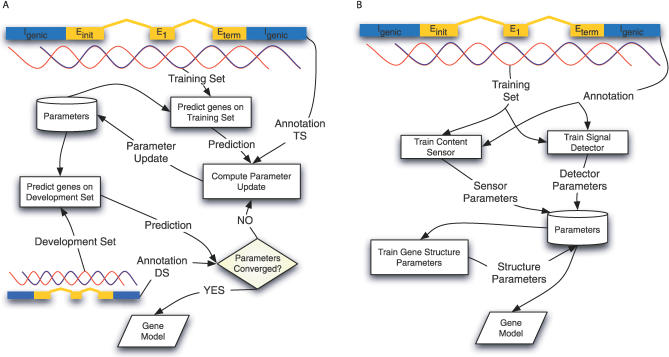
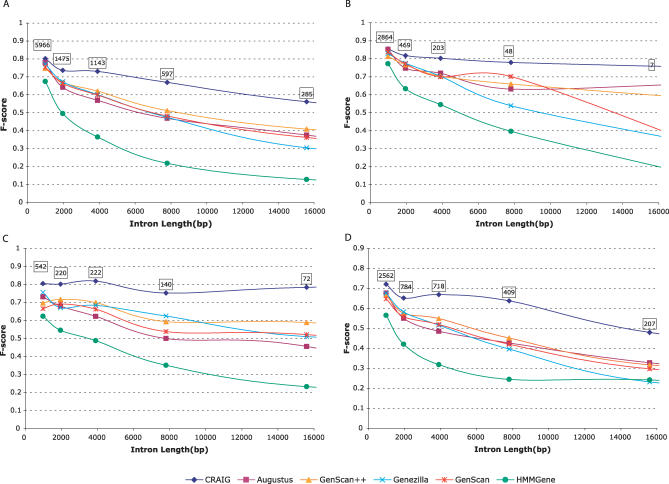
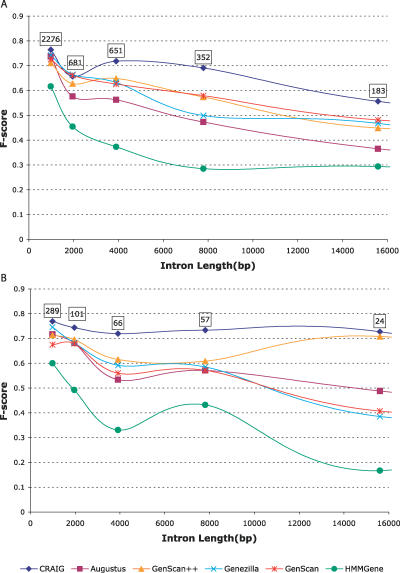
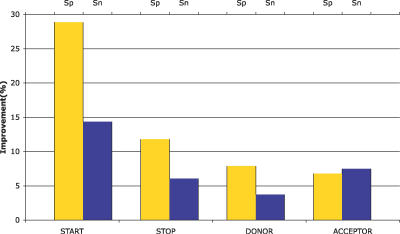
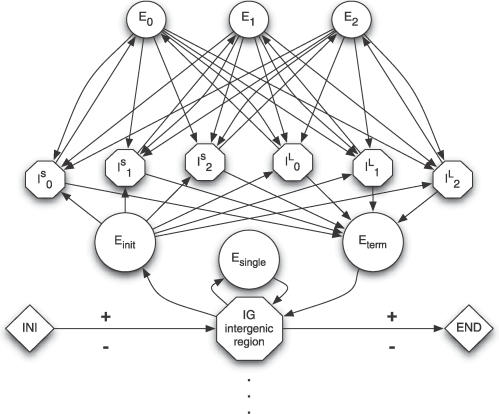
Most ab initio gene predictors use a probabilistic sequence model, typically a hidden Markov model, to combine separately trained models of genomic signals and content. By combining separate models of relevant genomic features, such gene predictors can exploit small training sets and incomplete annotations, and can be trained fairly efficiently. However, that type of piecewise training does not optimize prediction accuracy and has difficulty in accounting for statistical dependencies among different parts of the gene model. With genomic information being created at an ever-increasing rate, it is worth investigating alternative approaches in which many different types of genomic evidence, with complex statistical dependencies, can be integrated by discriminative learning to maximize annotation accuracy. Among discriminative learning methods, large-margin classifiers have become prominent because of the success of support vector machines (SVM) in many classification tasks. We describe CRAIG, a new program for ab initio gene prediction based on a conditional random field model with semi-Markov structure that is trained with an online large-margin algorithm related to multiclass SVMs. Our experiments on benchmark vertebrate datasets and on regions from the ENCODE project show significant improvements in prediction accuracy over published gene predictors that use intrinsic features only, particularly at the gene level and on genes with long introns.
Conflict of interest statement
Figures
References
-
- Burge CB, Karlin S. Finding the genes in genomic DNA. Curr Opin Struct Biol. 1998;8:346–354. - PubMed
-
- Stanke M, Waack S. Gene prediction with a hidden Markov model and a new intron submodel. Bioinformatics. 2003;19(Supplement 2):II215–II225. - PubMed
-
- Majoros WH, Pertea M, Salzberg SL. TigrScan and GlimmerHMM: Two open source ab initio eukaryotic genefinders. Bioinformatics. 2004;20:2878–2879. - PubMed
-
- Krogh A. Two methods for improving performance of an HMM and their application for gene finding. Proc Int Conf Intell Syst Mol Biol. 1997;5:179–186. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
