

Population genomics of the immune evasion (var) genes of Plasmodium falciparum
- PMID: 17367208
- PMCID: PMC1828697
- DOI: 10.1371/journal.ppat.0030034
Population genomics of the immune evasion (var) genes of Plasmodium falciparum
Abstract
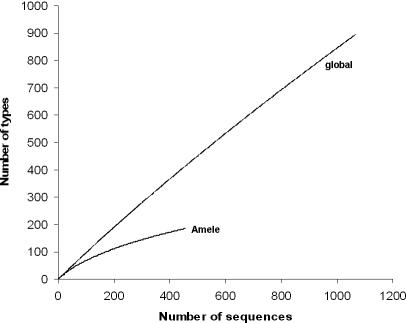
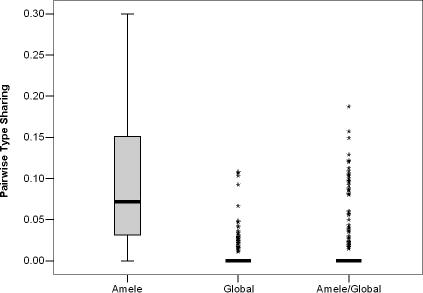
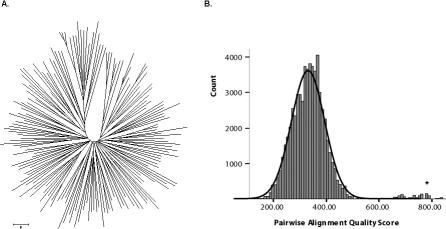
Var genes encode the major surface antigen (PfEMP1) of the blood stages of the human malaria parasite Plasmodium falciparum. Differential expression of up to 60 diverse var genes in each parasite genome underlies immune evasion. We compared the diversity of the DBLalpha domain of var genes sampled from 30 parasite isolates from a malaria endemic area of Papua New Guinea (PNG) and 59 from widespread geographic origins (global). Overall, we obtained over 8,000 quality-controlled DBLalpha sequences. Within our sampling frame, the global population had a total of 895 distinct DBLalpha "types" and negligible overlap among repertoires. This indicated that var gene diversity on a global scale is so immense that many genomes would need to be sequenced to capture its true extent. In contrast, we found a much lower diversity in PNG of 185 DBLalpha types, with an average of approximately 7% overlap among repertoires. While we identify marked geographic structuring, nearly 40% of types identified in PNG were also found in samples from different countries showing a cosmopolitan distribution for much of the diversity. We also present evidence to suggest that recombination plays a key role in maintaining the unprecedented levels of polymorphism found in these immune evasion genes. This population genomic framework provides a cost effective molecular epidemiological tool to rapidly explore the geographic diversity of var genes.
Conflict of interest statement
Figures
References
-
- Zambon MC. Epidemiology and pathogenesis of influenza. J Antimicrob Chemother. 1999;44(Suppl B):3–9. - PubMed
-
- Baruch DI, Pasloske BL, Singh HB, Bi X, Ma XC, et al. Cloning the P. falciparum gene encoding PfEMP1, a malarial variant antigen and adherence receptor on the surface of parasitized human erythrocytes. Cell. 1995;82:77–87. - PubMed
-
- Su XZ, Heatwole VM, Wertheimer SP, Guinet F, Herrfeldt J A, et al. The large diverse gene family var encodes proteins involved in cytoadherence and antigenic variation of Plasmodium falciparum-infected erythrocytes. Cell. 1995;82:89–100. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
