

MED: a new non-supervised gene prediction algorithm for bacterial and archaeal genomes
- PMID: 17367537
- PMCID: PMC1847833
- DOI: 10.1186/1471-2105-8-97
MED: a new non-supervised gene prediction algorithm for bacterial and archaeal genomes
Abstract
Background: Despite a remarkable success in the computational prediction of genes in Bacteria and Archaea, a lack of comprehensive understanding of prokaryotic gene structures prevents from further elucidation of differences among genomes. It continues to be interesting to develop new ab initio algorithms which not only accurately predict genes, but also facilitate comparative studies of prokaryotic genomes.
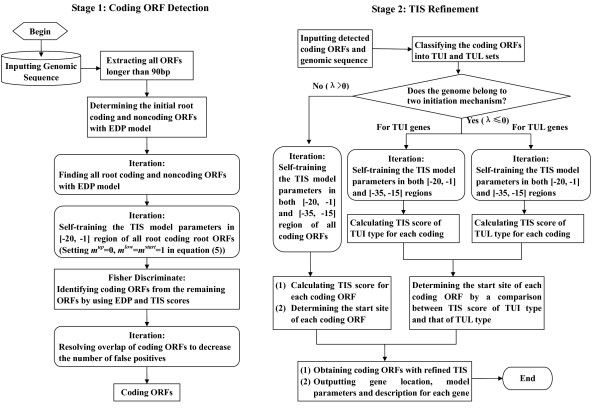
Results: This paper describes a new prokaryotic genefinding algorithm based on a comprehensive statistical model of protein coding Open Reading Frames (ORFs) and Translation Initiation Sites (TISs). The former is based on a linguistic "Entropy Density Profile" (EDP) model of coding DNA sequence and the latter comprises several relevant features related to the translation initiation. They are combined to form a so-called Multivariate Entropy Distance (MED) algorithm, MED 2.0, that incorporates several strategies in the iterative program. The iterations enable us to develop a non-supervised learning process and to obtain a set of genome-specific parameters for the gene structure, before making the prediction of genes.
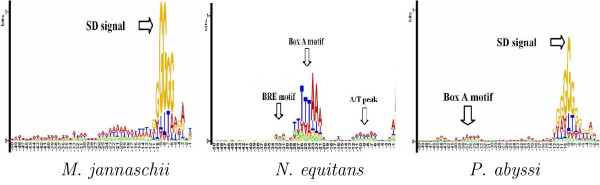
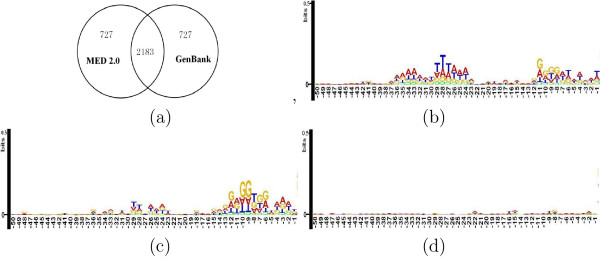
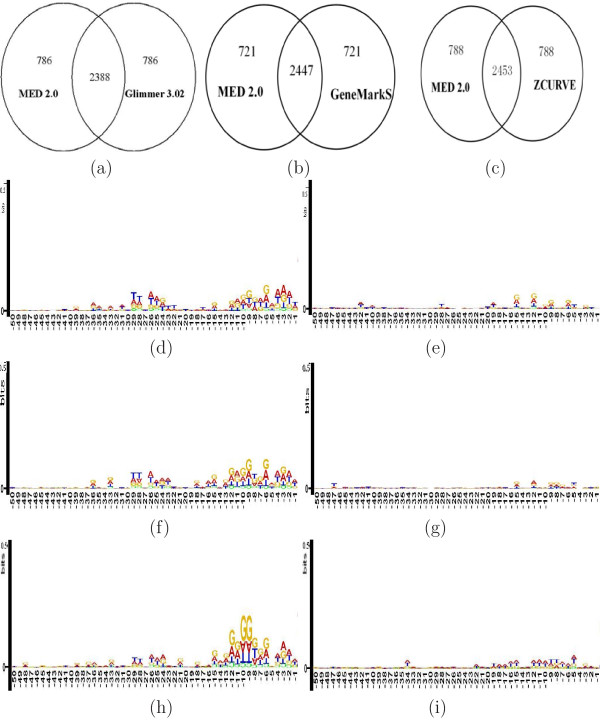
Conclusion: Results of extensive tests show that MED 2.0 achieves a competitive high performance in the gene prediction for both 5' and 3' end matches, compared to the current best prokaryotic gene finders. The advantage of the MED 2.0 is particularly evident for GC-rich genomes and archaeal genomes. Furthermore, the genome-specific parameters given by MED 2.0 match with the current understanding of prokaryotic genomes and may serve as tools for comparative genomic studies. In particular, MED 2.0 is shown to reveal divergent translation initiation mechanisms in archaeal genomes while making a more accurate prediction of TISs compared to the existing gene finders and the current GenBank annotation.
Figures
References
-
- Borodovsky M, Mclninch J. GENMARK: parallel gene recognition for both DNA strands. Comput Chem. 1993;17:123–133. doi: 10.1016/0097-8485(93)85004-V. - DOI
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
