

The original Lujan syndrome family has a novel missense mutation (p.N1007S) in the MED12 gene
- PMID: 17369503
- PMCID: PMC2597996
- DOI: 10.1136/jmg.2006.048637
The original Lujan syndrome family has a novel missense mutation (p.N1007S) in the MED12 gene
Abstract
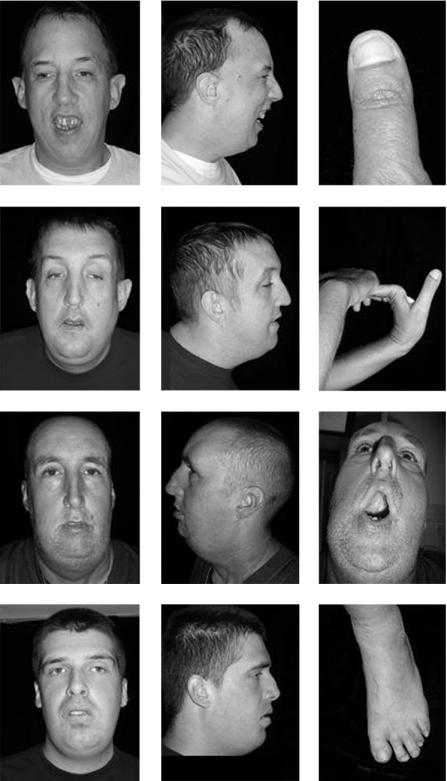
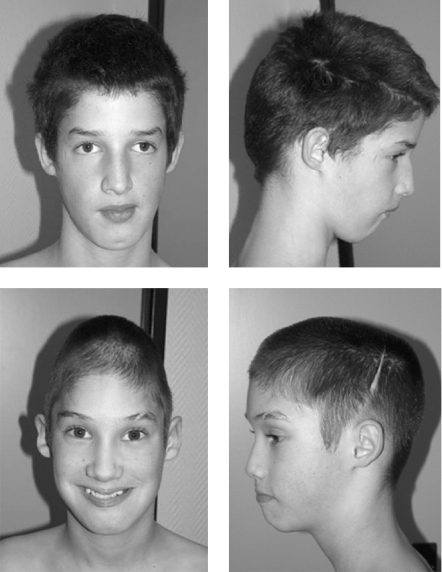
A novel missense mutation in the mediator of RNA polymerase II transcription subunit 12 (MED12) gene has been found in the original family with Lujan syndrome and in a second family (K9359) that was initially considered to have Opitz-Kaveggia (FG) syndrome. A different missense mutation in the MED12 gene has been reported previously in the original family with FG syndrome and in five other families with compatible clinical findings. Neither sequence alteration has been found in over 1400 control X chromosomes. Lujan (Lujan-Fryns) syndrome is characterised by tall stature with asthenic habitus, macrocephaly, a tall narrow face, maxillary hypoplasia, a high narrow palate with dental crowding, a small or receding chin, long hands with hyperextensible digits, hypernasal speech, hypotonia, mild-to-moderate mental retardation, behavioural aberrations and dysgenesis of the corpus callosum. Although Lujan syndrome has not been previously considered to be in the differential diagnosis of FG syndrome, there are some overlapping clinical manifestations. Specifically, these are dysgenesis of the corpus callosum, macrocephaly/relative macrocephaly, a tall forehead, hypotonia, mental retardation and behavioural disturbances. Thus, it seems that these two X-linked mental retardation syndromes are allelic, with mutations in the MED12 gene.
Conflict of interest statement
Competing interests: None.

References
-
- Stevenson R E. Splitting and lumping in the nosology of XLMR. Am J Med Genet (Semin Med Genet) 200097174–182. - PubMed
-
- Vits L, Van Camp G, Coucke P, Fransen E, De Boulle K, Reyniers E, Korn B, Poustka A, Wilson G, Schrander‐Stumpel C, Winter R M, Schwartz C, Willems P J. MASA syndrome is due to mutations in the neural cell adhesion gene L1CAM. Nat Genet 19947408–413. - PubMed
-
- Strømme P, Mangelsdorf M E, Shaw M A, Lower K M, Lewis S M, Bruyere H, Lutcherath V, Gedeon A K, Wallace R H, Scheffer I E, Turner G, Partington M, Frints S G, Fryns J P, Sutherland G R, Mulley J C, Gecz J. Mutations in the human ortholog of Aristaless cause X‐linked mental retardation and epilepsy. Nat Genet 200230441–445. - PubMed
-
- Bienvenu T, Poirier K, Friocourt G, Bahi N, Beaumont D, Fauchereau F, Ben Jeema L, Zemni R, Vinet M C, Francis F, Couvert P, Gomot M, Moraine C, van Bokhoven H, Kalscheuer V, Frints S, Geca J, Ohzaki K, Chaabouni H, Fryns J P, Desportes V, Beldjord C, Chelly J. ARX, a novel Prd‐class‐homeobox gene highly expressed in the telencephalon, is mutated in X‐linked mental retardation. Hum Mol Genet 200211981–991. - PubMed
-
- Kitamura K, Yanazawa M, Sugiyama N, Miura H, Iizuka‐Kogo A, Kusaka M, Omichi K, Suzuki R, Kato‐Fukui Y, Kamiirisa K, Matsuo M, Kamijo S, Kasahara M, Yoshioka H, Ogata T, Fukuda T, Kondo I, Kato M, Dobyns W B, Yokoyama M, Morohashi K. Mutation of ARX causes abnormal development of forebrain and testes in mice and X‐linked lissencephaly with abnormal genitalia in humans. Nat Genet 200232359–369. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases