

Mutation in the loop C-terminal to the cyclophilin A binding site of HIV-1 capsid protein disrupts proper virus assembly and infectivity
- PMID: 17371591
- PMCID: PMC1832212
- DOI: 10.1186/1742-4690-4-19
Mutation in the loop C-terminal to the cyclophilin A binding site of HIV-1 capsid protein disrupts proper virus assembly and infectivity
Abstract
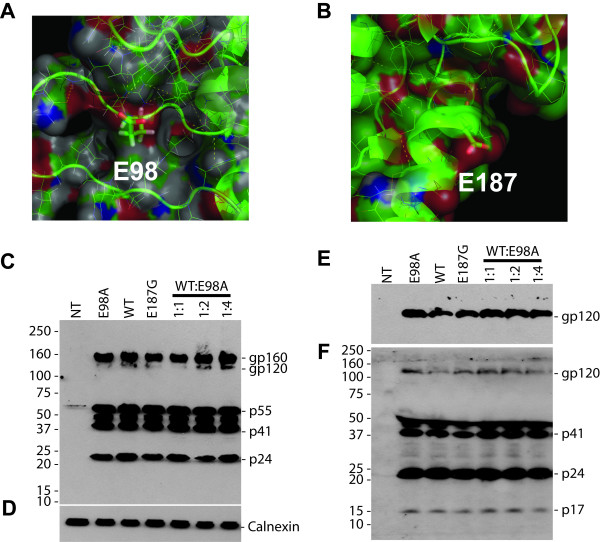
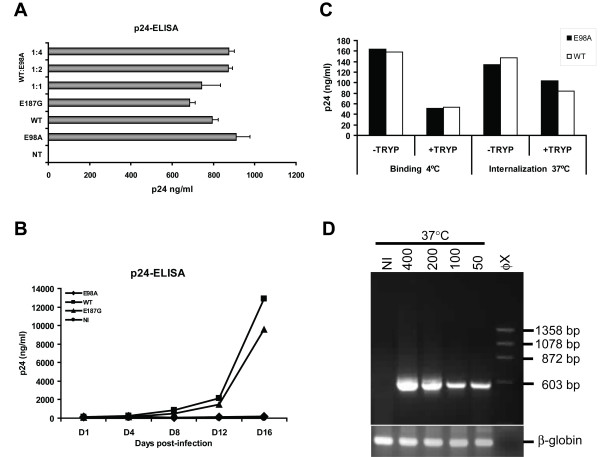
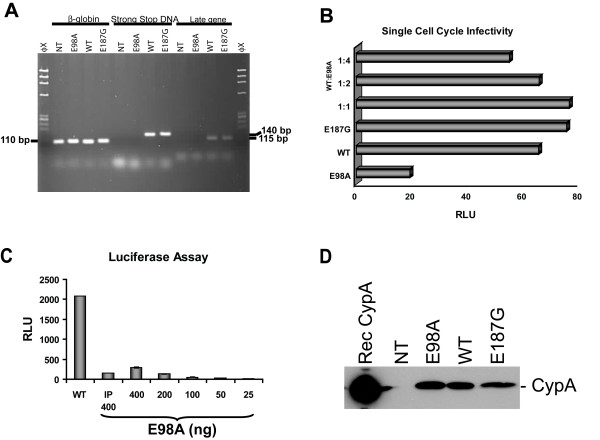
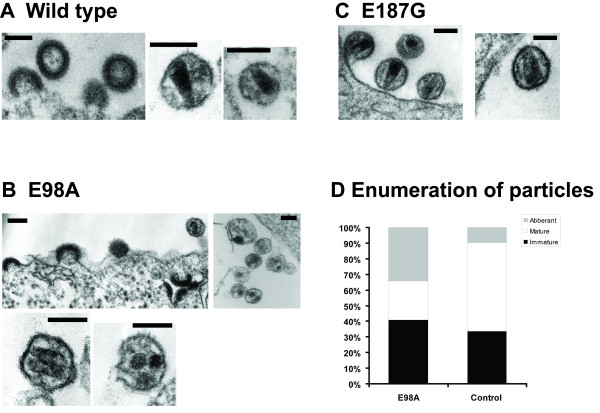
We have studied the effects associated with two single amino acid substitution mutations in HIV-1 capsid (CA), the E98A and E187G. Both amino acids are well conserved among all major HIV-1 subtypes. HIV-1 infectivity is critically dependent on proper CA cone formation and mutations in CA are lethal when they inhibit CA assembly by destabilizing the intra and/or inter molecular CA contacts, which ultimately abrogate viral replication. Glu98, which is located on a surface of a flexible cyclophilin A binding loop is not involved in any intra-molecular contacts with other CA residues. In contrast, Glu187 has extensive intra-molecular contacts with eight other CA residues. Additionally, Glu187 has been shown to form a salt-bridge with Arg18 of another N-terminal CA monomer in a N-C dimer. However, despite proper virus release, glycoprotein incorporation and Gag processing, electron microscopy analysis revealed that, in contrast to the E187G mutant, only the E98A particles had aberrant core morphology that resulted in loss of infectivity.
Figures
References
-
- Monaco-Malbet S, Berthet-Colominas C, Novelli A, Battai N, Piga N, Cheynet V, Mallet F, Cusack S. Mutual conformational adaptations in antigen and antibody upon complex formation between an Fab and HIV-1 capsid protein p24. Structure. 2000;8:1069–1077. doi: 10.1016/S0969-2126(00)00507-4. - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
