

Sonic hedgehog acts at multiple stages during pancreatic tumorigenesis
- PMID: 17372229
- PMCID: PMC1828712
- DOI: 10.1073/pnas.0701158104
Sonic hedgehog acts at multiple stages during pancreatic tumorigenesis
Abstract
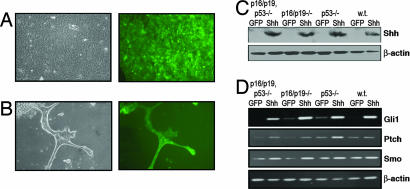
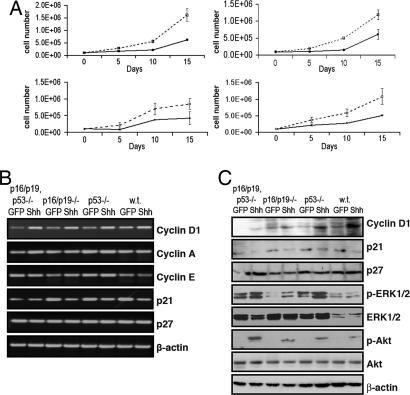
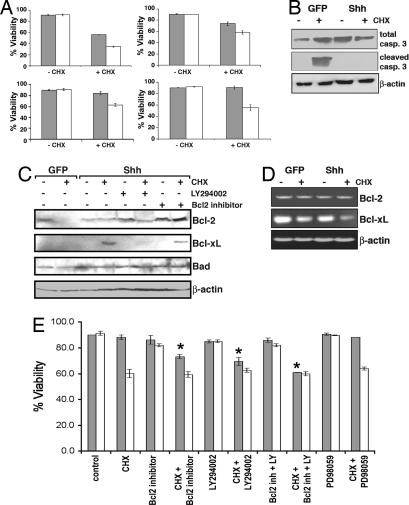
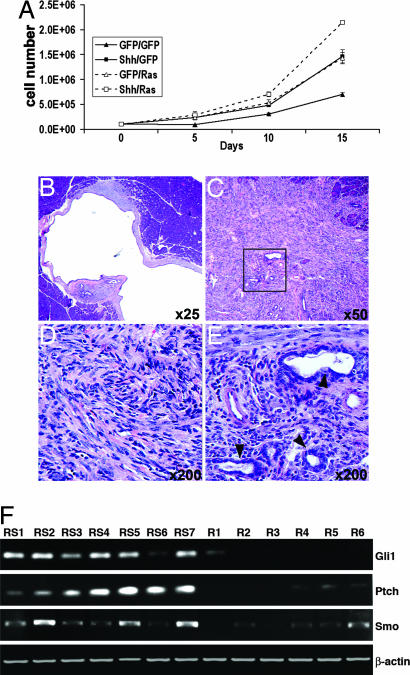
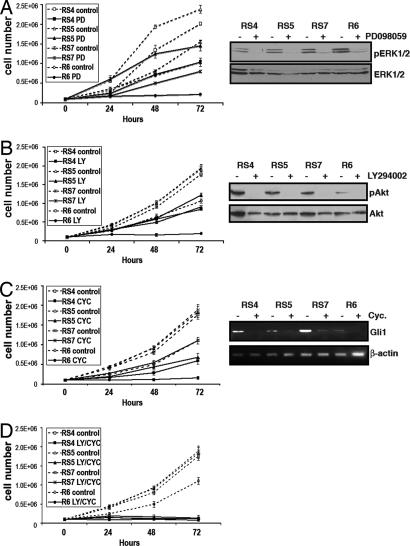
Activation of sonic hedgehog (Shh) signaling occurs in the majority of pancreatic ductal adenocarcinomas. Here we investigate the mechanisms by which Shh contributes to pancreatic tumorigenesis. We find that Shh expression enhances proliferation of pancreatic duct epithelial cells, potentially through the transcriptional regulation of the cell cycle regulators cyclin D1 and p21. We further show that Shh protects pancreatic duct epithelial cells from apoptosis through the activation of phosphatidylinositol 3-kinase signaling and the stabilization of Bcl-2 and Bcl-X(L). Significantly, Shh also cooperates with activated K-Ras to promote pancreatic tumor development. Finally, Shh signaling enhances K-Ras-induced pancreatic tumorigenesis by reducing the dependence of tumor cells on the sustained activation of the MAPK and phosphatidylinositol 3-kinase/Akt/mTOR signaling pathways. Thus, our data suggest that Shh signaling contributes to tumor initiation in the pancreas through at least two mechanisms and additionally enhances tumor cell resistance to therapeutic intervention. Collectively, our findings demonstrate crucial roles for Shh signaling in multiple stages of pancreatic carcinogenesis.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Shh signaling and pancreatic cancer: implications for therapy?Cell Cycle. 2007 Jul 1;6(13):1553-7. doi: 10.4161/cc.6.13.4467. Epub 2007 May 18. Cell Cycle. 2007. PMID: 17611415 Review.
-
Nuclear factor-kappaB contributes to hedgehog signaling pathway activation through sonic hedgehog induction in pancreatic cancer.Cancer Res. 2006 Jul 15;66(14):7041-9. doi: 10.1158/0008-5472.CAN-05-4588. Cancer Res. 2006. PMID: 16849549
-
In vitro modeling of human pancreatic duct epithelial cell transformation defines gene expression changes induced by K-ras oncogenic activation in pancreatic carcinogenesis.Cancer Res. 2005 Jun 15;65(12):5045-53. doi: 10.1158/0008-5472.CAN-04-3208. Cancer Res. 2005. PMID: 15958547
-
Sonic hedgehog paracrine signaling activates stromal cells to promote perineural invasion in pancreatic cancer.Clin Cancer Res. 2014 Aug 15;20(16):4326-38. doi: 10.1158/1078-0432.CCR-13-3426. Epub 2014 Jun 19. Clin Cancer Res. 2014. PMID: 24947933
-
Role of Sonic Hedgehog (Shh) Signaling in Bladder Cancer Stemness and Tumorigenesis.Curr Urol Rep. 2016 Feb;17(2):11. doi: 10.1007/s11934-015-0568-9. Curr Urol Rep. 2016. PMID: 26757905 Review.
Cited by
-
Codelivery of small molecule hedgehog inhibitor and miRNA for treating pancreatic cancer.Mol Pharm. 2015 Apr 6;12(4):1289-98. doi: 10.1021/mp500847s. Epub 2015 Feb 25. Mol Pharm. 2015. PMID: 25679326 Free PMC article.
-
Gli1 maintains cell survival by up-regulating IGFBP6 and Bcl-2 through promoter regions in parallel manner in pancreatic cancer cells.J Carcinog. 2009;8:13. doi: 10.4103/1477-3163.55429. J Carcinog. 2009. PMID: 19736394 Free PMC article.
-
Gli1 contributes to the invasiveness of pancreatic cancer through matrix metalloproteinase-9 activation.Cancer Sci. 2008 Jul;99(7):1377-84. doi: 10.1111/j.1349-7006.2008.00822.x. Epub 2008 Apr 11. Cancer Sci. 2008. PMID: 18410405 Free PMC article.
-
Expression and regulation of hedgehog signaling pathway in pancreatic cancer.Langenbecks Arch Surg. 2010 Jun;395(5):515-25. doi: 10.1007/s00423-009-0493-9. Epub 2009 Apr 25. Langenbecks Arch Surg. 2010. PMID: 19396459
-
Desire, disease, and the origins of the dopaminergic system.Schizophr Bull. 2008 Mar;34(2):212-9. doi: 10.1093/schbul/sbm170. Epub 2008 Feb 17. Schizophr Bull. 2008. PMID: 18283047 Free PMC article. Review.
References
-
- Jemal A, Murray T, Samuels A, Ghafoor A, Ward E, Thun MJ. CA Cancer J Clin. 2003;53:5–26. - PubMed
-
- Warshaw AL, Fernandez-del Castillo C. N Engl J Med. 1992;326:455–465. - PubMed
-
- Hruban RH, Goggins M, Parsons J, Kern SE. Clin Cancer Res. 2000;6:2969–2972. - PubMed
-
- Freeman M. Nature. 2000;408:313–319. - PubMed
-
- Pasca di Magliano M, Hebrok M. Nat Rev Cancer. 2003;3:903–911. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
