

Global identification and characterization of transcriptionally active regions in the rice genome
- PMID: 17372628
- PMCID: PMC1808428
- DOI: 10.1371/journal.pone.0000294
Global identification and characterization of transcriptionally active regions in the rice genome
Abstract
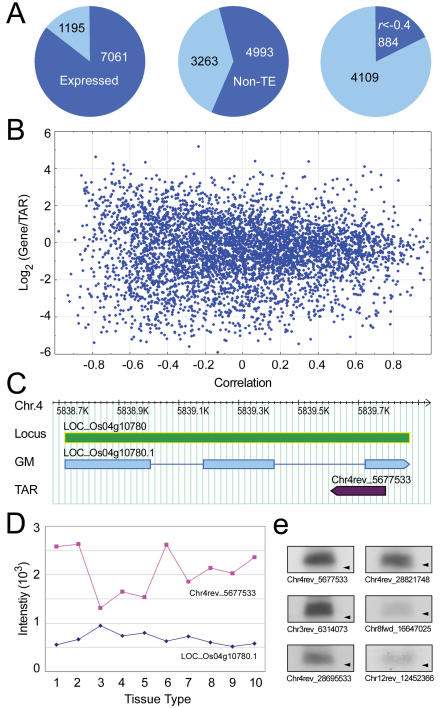
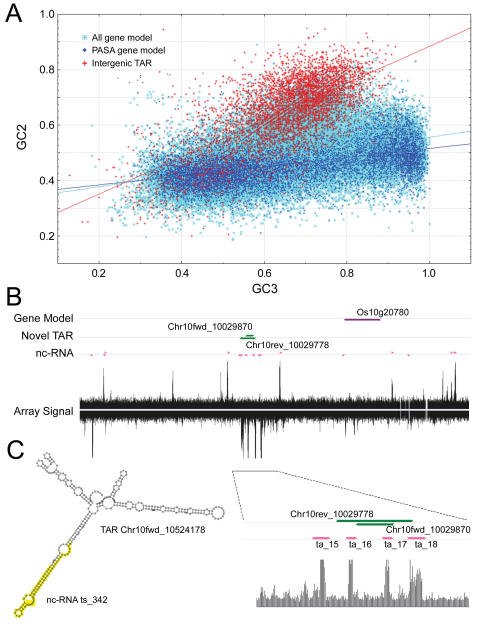
Genome tiling microarray studies have consistently documented rich transcriptional activity beyond the annotated genes. However, systematic characterization and transcriptional profiling of the putative novel transcripts on the genome scale are still lacking. We report here the identification of 25,352 and 27,744 transcriptionally active regions (TARs) not encoded by annotated exons in the rice (Oryza. sativa) subspecies japonica and indica, respectively. The non-exonic TARs account for approximately two thirds of the total TARs detected by tiling arrays and represent transcripts likely conserved between japonica and indica. Transcription of 21,018 (83%) japonica non-exonic TARs was verified through expression profiling in 10 tissue types using a re-array in which annotated genes and TARs were each represented by five independent probes. Subsequent analyses indicate that about 80% of the japonica TARs that were not assigned to annotated exons can be assigned to various putatively functional or structural elements of the rice genome, including splice variants, uncharacterized portions of incompletely annotated genes, antisense transcripts, duplicated gene fragments, and potential non-coding RNAs. These results provide a systematic characterization of non-exonic transcripts in rice and thus expand the current view of the complexity and dynamics of the rice transcriptome.
Conflict of interest statement
Figures





Similar articles
-
Tiling microarray analysis of rice chromosome 10 to identify the transcriptome and relate its expression to chromosomal architecture.Genome Biol. 2005;6(6):R52. doi: 10.1186/gb-2005-6-6-r52. Epub 2005 May 27. Genome Biol. 2005. PMID: 15960804 Free PMC article.
-
Antisense transcripts with rice full-length cDNAs.Genome Biol. 2003;5(1):R5. doi: 10.1186/gb-2003-5-1-r5. Epub 2003 Dec 11. Genome Biol. 2003. PMID: 14709177 Free PMC article.
-
A pilot study of transcription unit analysis in rice using oligonucleotide tiling-path microarray.Plant Mol Biol. 2005 Sep;59(1):137-49. doi: 10.1007/s11103-005-6164-5. Plant Mol Biol. 2005. PMID: 16217608
-
Transcribed dark matter: meaning or myth?Hum Mol Genet. 2010 Oct 15;19(R2):R162-8. doi: 10.1093/hmg/ddq362. Epub 2010 Aug 25. Hum Mol Genet. 2010. PMID: 20798109 Free PMC article. Review.
-
[DNA arrays: a general overview and specific applications].Med Clin (Barc). 2008 Apr 12;130(13):504-9. doi: 10.1157/13119504. Med Clin (Barc). 2008. PMID: 18423170 Review. Spanish.
Cited by
-
Expression dynamics of metabolic and regulatory components across stages of panicle and seed development in indica rice.Funct Integr Genomics. 2012 Jun;12(2):229-48. doi: 10.1007/s10142-012-0274-3. Epub 2012 Mar 31. Funct Integr Genomics. 2012. PMID: 22466020
-
Transcriptional profiling of long non-coding RNAs regulating fruit cracking in Punica granatum L. under bagging.Front Plant Sci. 2022 Oct 11;13:943547. doi: 10.3389/fpls.2022.943547. eCollection 2022. Front Plant Sci. 2022. PMID: 36304394 Free PMC article.
-
Identification and Analysis of Long Non-Coding RNAs Related to UV-B-Induced Anthocyanin Biosynthesis During Blood-Fleshed Peach (Prunus persica) Ripening.Front Genet. 2022 Aug 9;13:932207. doi: 10.3389/fgene.2022.932207. eCollection 2022. Front Genet. 2022. PMID: 36017497 Free PMC article.
-
AGRONOMICS1: a new resource for Arabidopsis transcriptome profiling.Plant Physiol. 2010 Feb;152(2):487-99. doi: 10.1104/pp.109.150185. Epub 2009 Dec 23. Plant Physiol. 2010. PMID: 20032078 Free PMC article.
-
High-resolution mapping of epigenetic modifications of the rice genome uncovers interplay between DNA methylation, histone methylation, and gene expression.Plant Cell. 2008 Feb;20(2):259-76. doi: 10.1105/tpc.107.056879. Epub 2008 Feb 8. Plant Cell. 2008. PMID: 18263775 Free PMC article.
References
-
- Goff SA, Ricke D, Lan TH, Presting G, Wang R, et al. A draft sequence of the rice genome (Oryza sativa L. ssp japonica). Science. 2002;296:92–100. - PubMed
-
- Yu J, Hu S, Wang J, Wong GK, Li S, et al. A draft sequence of the rice genome (Oryza sativa L. ssp. indica). Science. 2002;296:79–92. - PubMed
-
- Sasaki T, Matsumoto T, Yamamoto K, Sakata K, Baba T, et al. The genome sequence and structure of rice chromosome 1. Nature. 2002;420:312–316. - PubMed
-
- Feng Q, Zhang Y, Hao P, Wang S, Fu G, et al. Sequence and analysis of rice chromosome 4. Nature. 2002;420:316–320. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
