

Mechanism of membrane redistribution of protein kinase C by its ATP-competitive inhibitors
- PMID: 17373912
- PMCID: PMC1904528
- DOI: 10.1042/BJ20070299
Mechanism of membrane redistribution of protein kinase C by its ATP-competitive inhibitors
Abstract
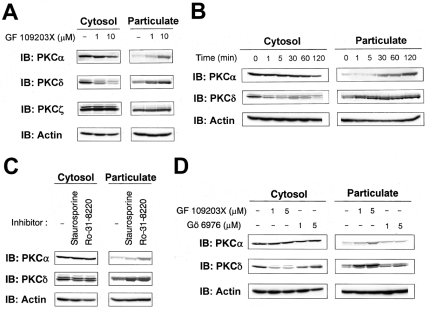
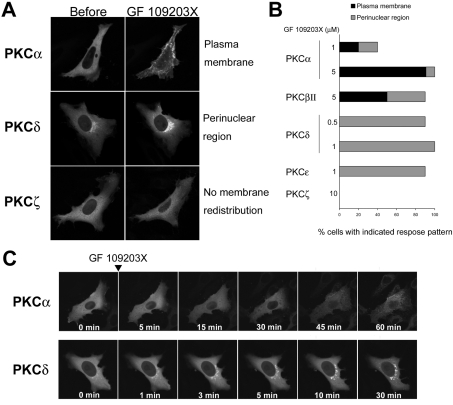
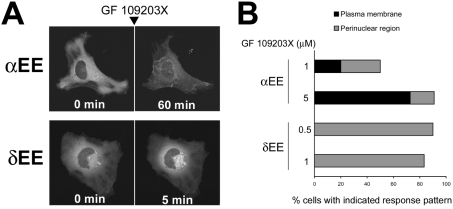
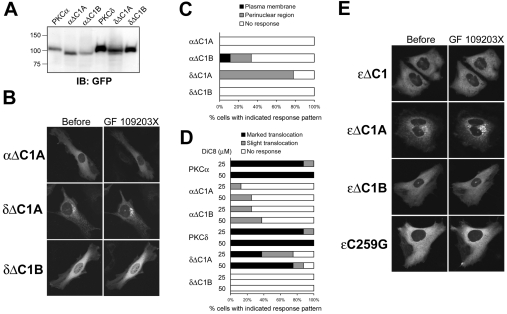
ATP-competitive inhibitors of PKC (protein kinase C) such as the bisindolylmaleimide GF 109203X, which interact with the ATP-binding site in the PKC molecule, have also been shown to affect several redistribution events of PKC. However, the reason why these inhibitors affect the redistribution is still controversial. In the present study, using immunoblot analysis and GFP (green fluorescent protein)-tagged PKC, we showed that, at commonly used concentrations, these ATP-competitive inhibitors alone induced redistribution of DAG (diacylglycerol)-sensitive PKCalpha, PKCbetaII, PKCdelta and PKCepsilon, but not atypical PKCzeta, to the endomembrane or the plasma membrane. Studies with deletion and point mutants showed that the DAG-sensitive C1 domain of PKC was required for membrane redistribution by these inhibitors. Furthermore, membrane redistribution was prevented by the aminosteroid PLC (phospholipase C) inhibitor U-73122, although an ATP-competitive inhibitor had no significant effect on acute DAG generation. Immunoblot analysis showed that an ATP-competitive inhibitor enhanced cell-permeable DAG analogue- or phorbol-ester-induced translocation of endogenous PKC. Furthermore, these inhibitors also enhanced [3H]phorbol 12,13-dibutyrate binding to the cytosolic fractions from PKCalpha-GFP-overexpressing cells. These results clearly demonstrate that ATP-competitive inhibitors cause redistribution of DAG-sensitive PKCs to membranes containing endogenous DAG by altering the DAG sensitivity of PKC and support the idea that the inhibitors destabilize the closed conformation of PKC and make the C1 domain accessible to DAG. Most importantly, our findings provide novel insights for the interpretation of studies using ATP-competitive inhibitors, and, especially, suggest caution about the interpretation of the relationship between the redistribution and kinase activity of PKC.
Figures
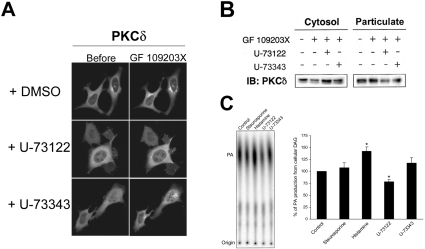
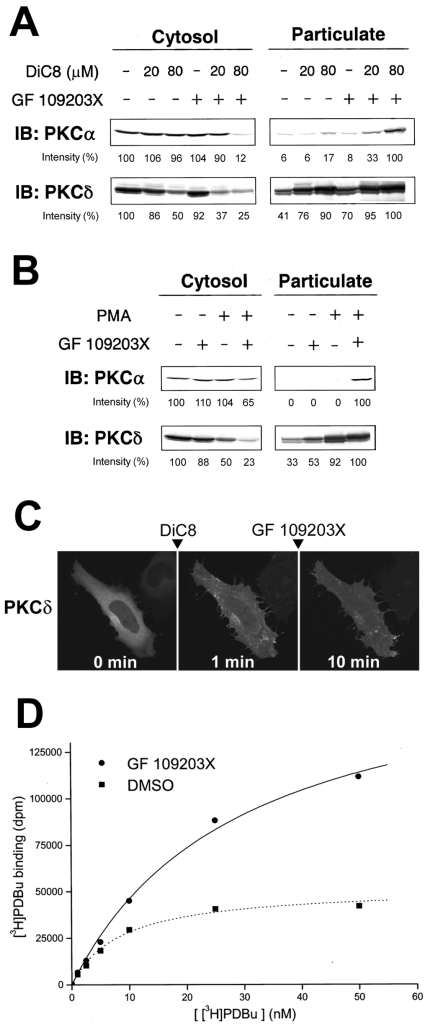
References
-
- Newton A. C. Regulation of protein kinase C. Curr. Opin. Cell Biol. 1997;9:161–167. - PubMed
-
- Nishizuka Y. Intracellular signaling by hydrolysis of phospholipids and activation of protein kinase C. Science. 1992;258:607–614. - PubMed
-
- Farrar W. L., Anderson W. B. Interleukin-2 stimulates association of protein kinase C with plasma membrane. Nature. 1985;315:233–235. - PubMed
-
- Oancea E., Meyer T. Protein kinase C as a molecular machine for decoding calcium and diacylglycerol signals. Cell. 1998;95:307–318. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources