

Expression and characterization of mutations in human very long-chain acyl-CoA dehydrogenase using a prokaryotic system
- PMID: 17374501
- PMCID: PMC2702680
- DOI: 10.1016/j.ymgme.2007.01.013
Expression and characterization of mutations in human very long-chain acyl-CoA dehydrogenase using a prokaryotic system
Abstract
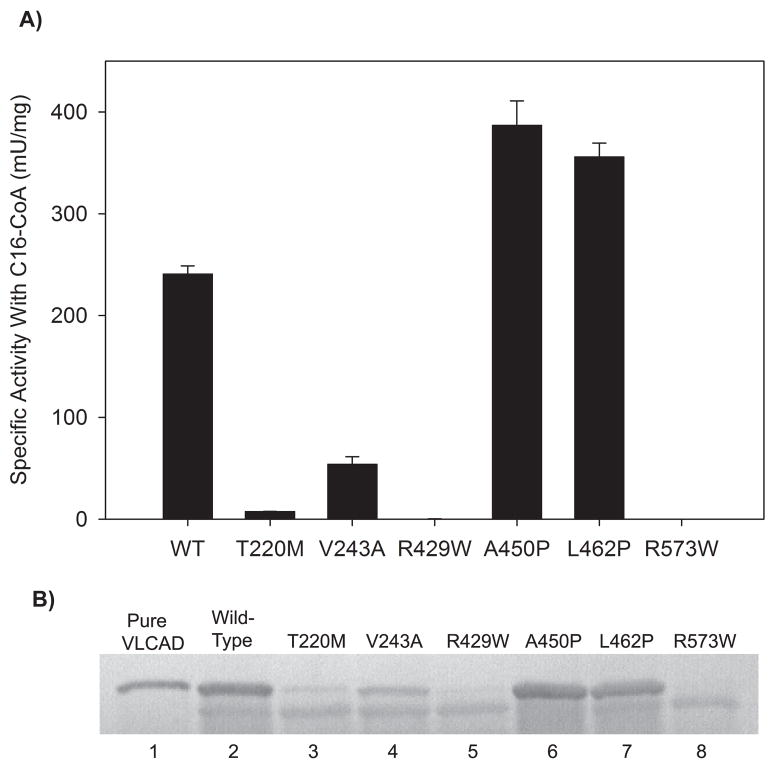
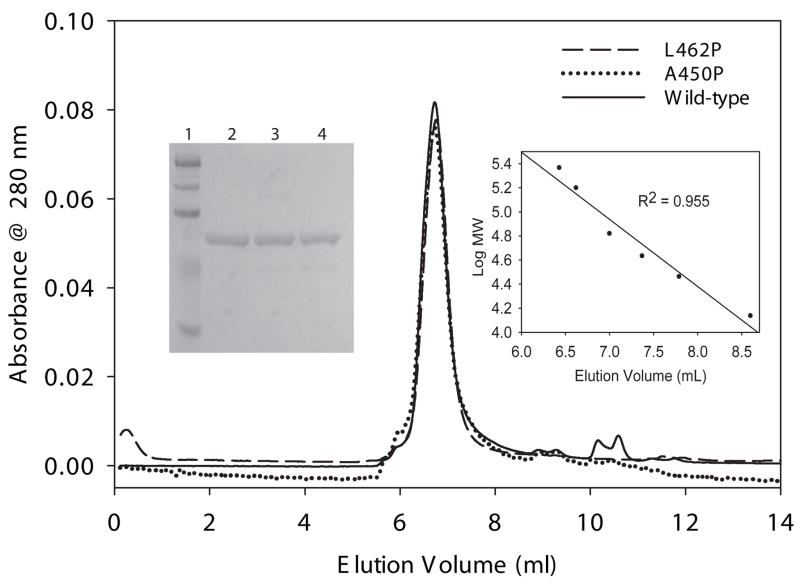
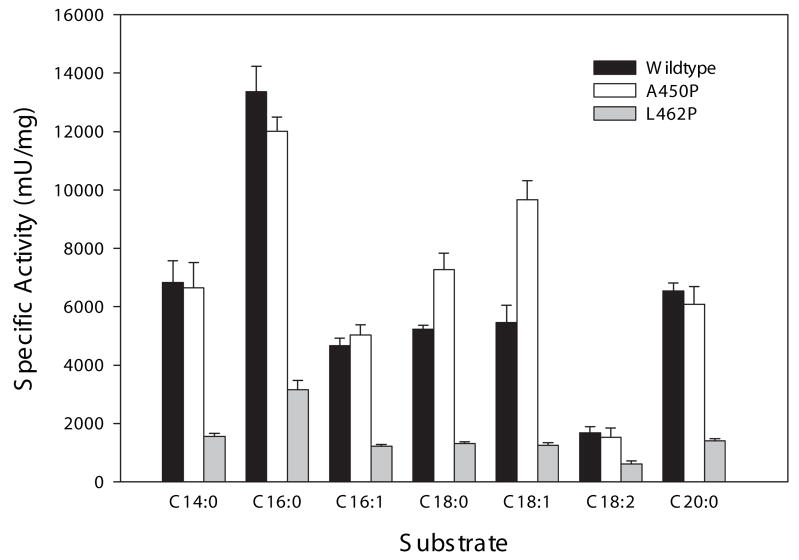
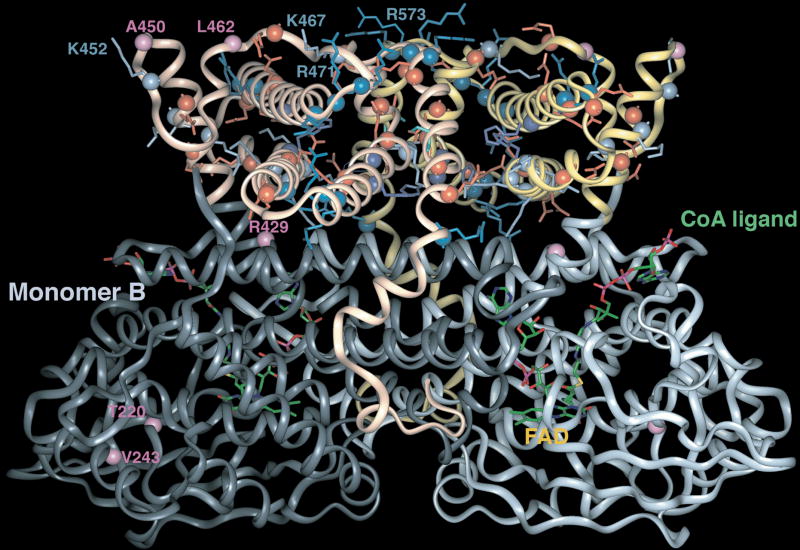
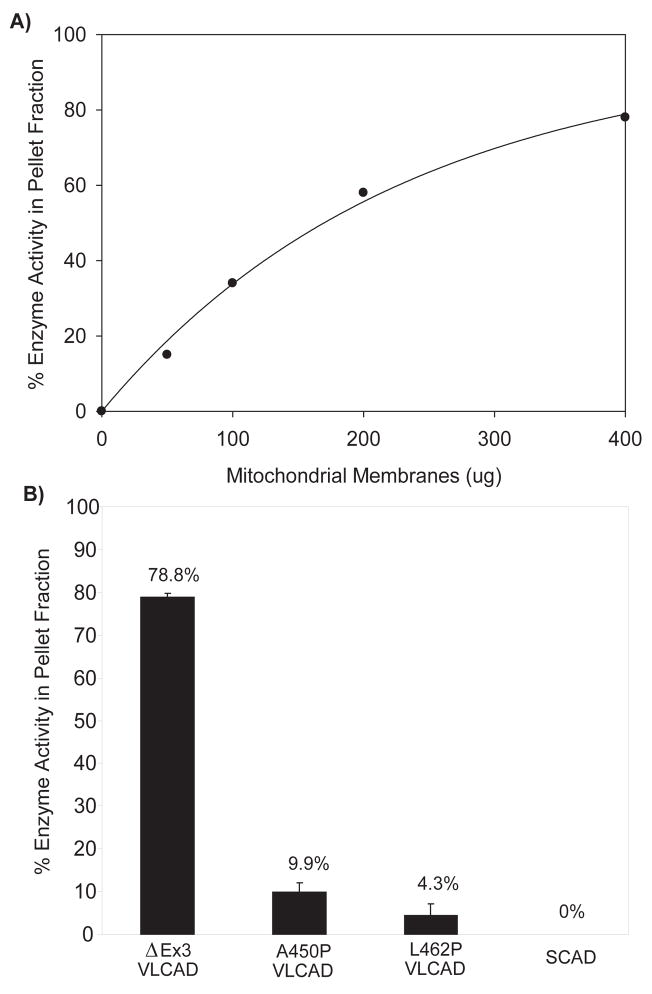
Very long-chain acyl-CoA dehydrogenase (VLCAD) catalyzes the first enzymatic step in the mitochondrial beta-oxidation of fatty acids 14-20 carbons in length. More than 100 cases of VLCAD deficiency have been reported with the disease varying from a severe, often fatal neonatal form to a mild adult-onset form. VLCAD is distinguished from matrix-soluble acyl-CoA dehydrogenases by its unique C-terminal domain, homodimeric structure, and localization to the inner mitochondrial membrane. We have for the first time expressed and purified VLCAD using a bacterial system. Recombinant VLCAD had similar biochemical properties to those reported for native VLCAD and the bacterial system was used to study six previously described disease-causing missense mutations including the two most common mild mutations (T220M, V243A), a mutation leading to the severe disease phenotype (R429W), and three mutations in the C-terminal domain (A450P, L462P, and R573W). Of particular interest was the finding that the A450P and L462P bacterial extracts had normal or increased amounts of VLCAD antigen and activity. In the pure form L462P had roughly 30% of wild-type activity while A450P was normal. Using computer modeling both mutations were mapped to a predicted charged surface of VLCAD that we postulate interacts with the mitochondrial membrane. In a membrane pull down assay both mutants showed greatly reduced mitochondrial membrane association, suggesting a mechanism for the disease in these patients. In summary, the bacterial expression system developed here will significantly advance our understanding of both the clinical aspects of VLCAD deficiency and the basic biochemistry of the enzyme.
Figures
Similar articles
-
Structural basis for defective membrane targeting of mutant enzyme in human VLCAD deficiency.Nat Commun. 2022 Jun 27;13(1):3669. doi: 10.1038/s41467-022-31466-2. Nat Commun. 2022. PMID: 35760926 Free PMC article.
-
A heterozygous missense mutation in adolescent-onset very long-chain acyl-CoA dehydrogenase deficiency with exercise-induced rhabdomyolysis.Tohoku J Exp Med. 2015 Apr;235(4):305-10. doi: 10.1620/tjem.235.305. Tohoku J Exp Med. 2015. PMID: 25843429
-
Relationship between structure and substrate-chain-length specificity of mitochondrial very-long-chain acyl-coenzyme A dehydrogenase.Eur J Biochem. 1998 Nov 1;257(3):592-8. doi: 10.1046/j.1432-1327.1998.2570592.x. Eur J Biochem. 1998. PMID: 9839948
-
Very long-chain acyl-CoA dehydrogenase (VLCAD-) deficiency-studies on treatment effects and long-term outcomes in mouse models.J Inherit Metab Dis. 2017 May;40(3):317-323. doi: 10.1007/s10545-017-0016-8. Epub 2017 Feb 28. J Inherit Metab Dis. 2017. PMID: 28247148 Review.
-
Management and diagnosis of mitochondrial fatty acid oxidation disorders: focus on very-long-chain acyl-CoA dehydrogenase deficiency.J Hum Genet. 2019 Feb;64(2):73-85. doi: 10.1038/s10038-018-0527-7. Epub 2018 Nov 6. J Hum Genet. 2019. PMID: 30401918 Review.
Cited by
-
Genetic basis for correction of very-long-chain acyl-coenzyme A dehydrogenase deficiency by bezafibrate in patient fibroblasts: toward a genotype-based therapy.Am J Hum Genet. 2007 Dec;81(6):1133-43. doi: 10.1086/522375. Epub 2007 Oct 29. Am J Hum Genet. 2007. PMID: 17999356 Free PMC article.
-
Mitochondrial energetics is impaired in very long-chain acyl-CoA dehydrogenase deficiency and can be rescued by treatment with mitochondria-targeted electron scavengers.Hum Mol Genet. 2019 Mar 15;28(6):928-941. doi: 10.1093/hmg/ddy403. Hum Mol Genet. 2019. PMID: 30445591 Free PMC article.
-
Mitochondrial fatty acid oxidation disorders: clinical presentation of long-chain fatty acid oxidation defects before and after newborn screening.J Inherit Metab Dis. 2010 Oct;33(5):527-32. doi: 10.1007/s10545-010-9090-x. Epub 2010 May 7. J Inherit Metab Dis. 2010. PMID: 20449660 Review.
-
Structural basis for defective membrane targeting of mutant enzyme in human VLCAD deficiency.Nat Commun. 2022 Jun 27;13(1):3669. doi: 10.1038/s41467-022-31466-2. Nat Commun. 2022. PMID: 35760926 Free PMC article.
-
Successful Treatment of Cardiomyopathy due to Very Long-Chain Acyl-CoA Dehydrogenase Deficiency: First Case Report from Oman with Literature Review.Oman Med J. 2013 Sep;28(5):354-6. doi: 10.5001/omj.2013.101. Oman Med J. 2013. PMID: 24044064 Free PMC article.
References
-
- Gregersen N, Andresen BS, Corydon MJ, Corydon TJ, Olsen RK, Bolund L, Bross P. Mutation analysis in mitochondrial fatty acid oxidation defects: Exemplified by acyl-CoA dehydrogenase deficiencies, with special focus on genotype-phenotype relationship. Hum Mutat. 2001;18:169–89. - PubMed
-
- Sweetman L, Williams JD. Branched chain organic acidurias. In: Scriver C, Beaudet AL, Sly W, Valle D, editors. The Metabolic and Molecular Basis of Inherited Disease. 8. New York: McGraw-Hill; 2001. pp. 2125–64.
-
- Wanders RJA, Vreken P, Den Boer MEJ, Wijburg FA, Van Gennip AH, IJlst L. Disorders of mitochondrial fatty acyl-CoA β–oxidation. J Inher Metab Dis. 1999;22:442–87. - PubMed
-
- Vockley J, Singh RH, Whiteman DA. Diagnosis and management of defects of mitochondrial beta-oxidation. Curr Opin Clin Nutr Metab Care. 2002;5:601–9. - PubMed
-
- Andresen BS, Olpin S, Poorthuis Bjhm, Scholte HR, Vianey-Saban C, Wanders R, Ijlst L, Morris A, Pourfarzam M, Bartlett K, Baumgartner ER, deKlerk JBC, Schroeder LD, Corydon TJ, Lund H, Winter V, Bross P, Bolund L, Gregersen N. Clear correlation of genotype with disease phenotype in very-long-chain acyl-CoA dehydrogenase deficiency. Am J Hum Genet. 1999;64:479–94. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
