

Random screening for dominant-negative mutants of the cytomegalovirus nuclear egress protein M50
- PMID: 17376929
- PMCID: PMC1900260
- DOI: 10.1128/JVI.02796-06
Random screening for dominant-negative mutants of the cytomegalovirus nuclear egress protein M50
Abstract
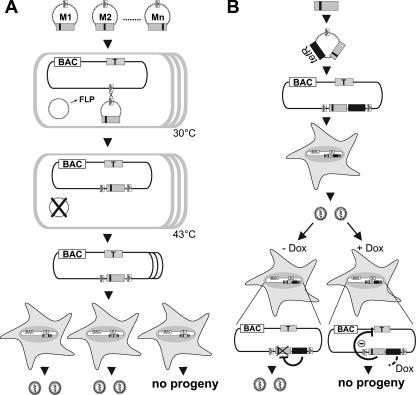
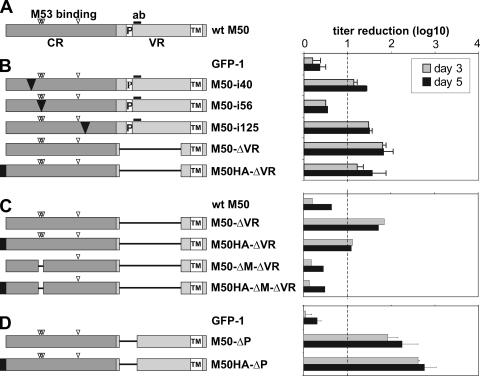
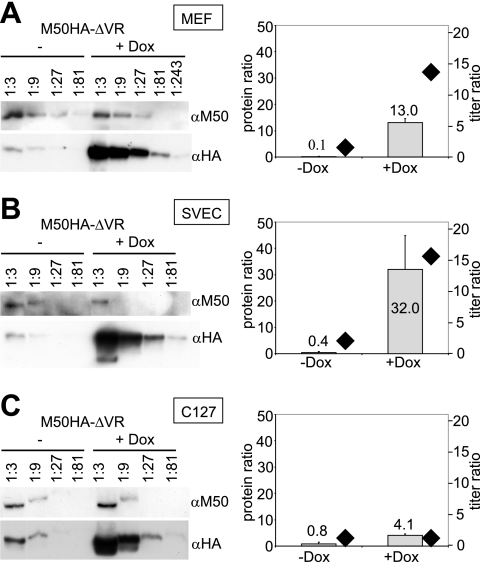
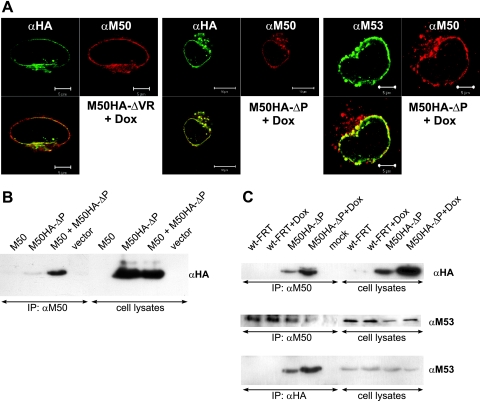
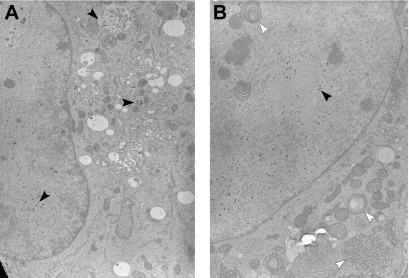
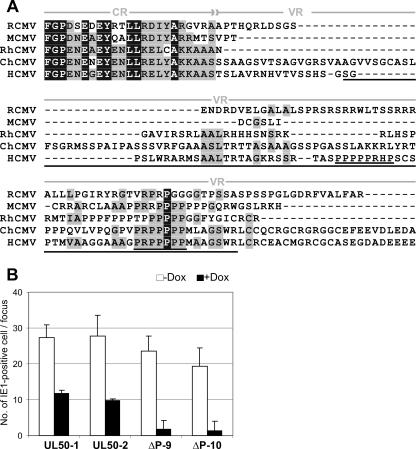
Inactivation of gene products by dominant-negative (DN) mutants is a powerful tool to assign functions to proteins. Here, we present a two-step procedure to establish a random screen for DN alleles, using the essential murine cytomegalovirus gene M50 as an example. First, loss-of-function mutants from a linker-scanning library were tested for inhibition of virus reconstitution with the help of FLP-mediated ectopic insertion of the mutants into the viral genome. Second, DN candidates were confirmed by conditional expression of the inhibitory proteins in the virus context. This allowed the quantification of the inhibitory effect, the identification of the morphogenesis block, and the construction of DN mutants with improved activity. Based on these observations a DN mutant of the homologous gene (UL50) in human cytomegalovirus was predicted and constructed. Our data suggest that a proline-rich sequence motif in the variable region of M50/UL50 represents a new functional site which is essential for nuclear egress of cytomegalovirus capsids.
Figures

References
-
- Adler, B., L. Scrivano, Z. Ruzsics, B. Rupp, C. Sinzger, and U. Koszinowski. 2006. Role of human cytomegalovirus UL131A in cell type-specific virus entry and release. J. Gen. Virol. 87:2451-2460. - PubMed
-
- Baltimore, D. 1988. Gene therapy. Intracellular immunization. Nature 335:395-396. - PubMed
-
- Brune, W., C. Menard, J. Heesemann, and U. H. Koszinowski. 2001. A ribonucleotide reductase homolog of cytomegalovirus and endothelial cell tropism. Science 291:303-305. - PubMed
-
- Brune, W., C. Menard, U. Hobom, S. Odenbreit, M. Messerle, and U. H. Koszinowski. 1999. Rapid identification of essential and nonessential herpesvirus genes by direct transposon mutagenesis. Nat. Biotechnol. 17:360-364. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
