

Scoring predictive models using a reduced representation of proteins: model and energy definition
- PMID: 17378941
- PMCID: PMC1854906
- DOI: 10.1186/1472-6807-7-15
Scoring predictive models using a reduced representation of proteins: model and energy definition
Abstract
Background: Reduced representations of proteins have been playing a keyrole in the study of protein folding. Many such models are available, with different representation detail. Although the usefulness of many such models for structural bioinformatics applications has been demonstrated in recent years, there are few intermediate resolution models endowed with an energy model capable, for instance, of detecting native or native-like structures among decoy sets. The aim of the present work is to provide a discrete empirical potential for a reduced protein model termed here PC2CA, because it employs a PseudoCovalent structure with only 2 Centers of interactions per Amino acid, suitable for protein model quality assessment.
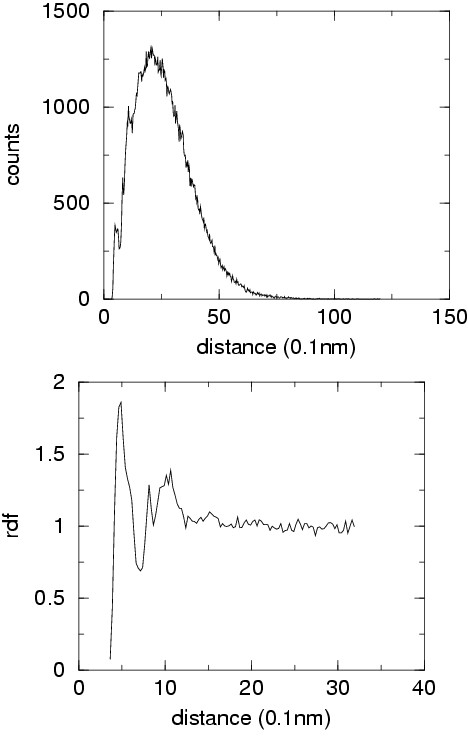
Results: All protein structures in the set top500H have been converted in reduced form. The distribution of pseudobonds, pseudoangle, pseudodihedrals and distances between centers of interactions have been converted into potentials of mean force. A suitable reference distribution has been defined for non-bonded interactions which takes into account excluded volume effects and protein finite size. The correlation between adjacent main chain pseudodihedrals has been converted in an additional energetic term which is able to account for cooperative effects in secondary structure elements. Local energy surface exploration is performed in order to increase the robustness of the energy function.
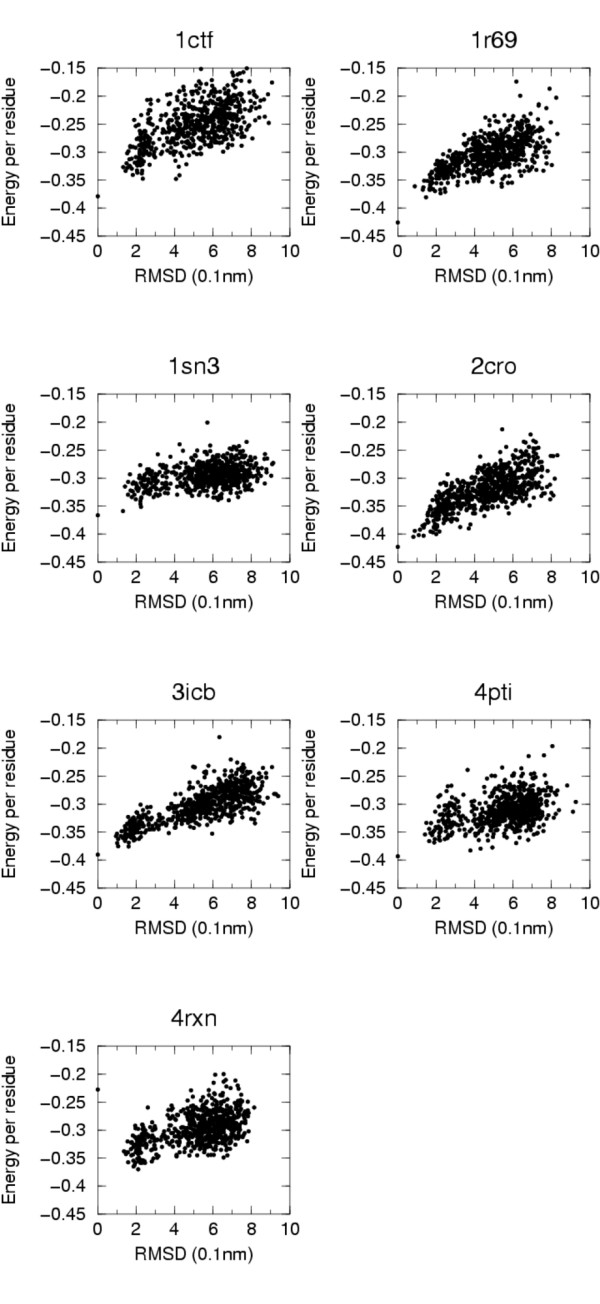
Conclusion: The model and the energy definition proposed have been tested on all the multiple decoys' sets in the Decoys'R'us database. The energetic model is able to recognize, for almost all sets, native-like structures (RMSD less than 2.0 A). These results and those obtained in the blind CASP7 quality assessment experiment suggest that the model compares well with scoring potentials with finer granularity and could be useful for fast exploration of conformational space. Parameters are available at the url: http://www.dstb.uniud.it/~ffogolari/download/.
Figures

Similar articles
-
Distinguishing native conformations of proteins from decoys with an effective free energy estimator based on the OPLS all-atom force field and the Surface Generalized Born solvent model.Proteins. 2002 Aug 1;48(2):404-22. doi: 10.1002/prot.10171. Proteins. 2002. PMID: 12112706
-
Protein structure evaluation using an all-atom energy based empirical scoring function.J Biomol Struct Dyn. 2006 Feb;23(4):385-406. doi: 10.1080/07391102.2006.10531234. J Biomol Struct Dyn. 2006. PMID: 16363875
-
A reduced protein model with accurate native-structure identification ability.Proteins. 2003 Dec 1;53(4):889-907. doi: 10.1002/prot.10498. Proteins. 2003. PMID: 14635131
-
Protein modeling with reduced representation: statistical potentials and protein folding mechanism.Acta Biochim Pol. 2005;52(4):741-8. Epub 2005 May 31. Acta Biochim Pol. 2005. PMID: 15933762 Review.
-
Theory of protein folding: the energy landscape perspective.Annu Rev Phys Chem. 1997;48:545-600. doi: 10.1146/annurev.physchem.48.1.545. Annu Rev Phys Chem. 1997. PMID: 9348663 Review.
Cited by
-
Selective refinement and selection of near-native models in protein structure prediction.Proteins. 2015 Oct;83(10):1823-35. doi: 10.1002/prot.24866. Epub 2015 Aug 12. Proteins. 2015. PMID: 26214389 Free PMC article.
-
Four-body atomic potential for modeling protein-ligand binding affinity: application to enzyme-inhibitor binding energy prediction.BMC Struct Biol. 2013;13 Suppl 1(Suppl 1):S1. doi: 10.1186/1472-6807-13-S1-S1. Epub 2013 Nov 8. BMC Struct Biol. 2013. PMID: 24564918 Free PMC article.
-
A pairwise residue contact area-based mean force potential for discrimination of native protein structure.BMC Bioinformatics. 2010 Jan 9;11:16. doi: 10.1186/1471-2105-11-16. BMC Bioinformatics. 2010. PMID: 20064218 Free PMC article.
-
NCACO-score: an effective main-chain dependent scoring function for structure modeling.BMC Bioinformatics. 2011 May 26;12:208. doi: 10.1186/1471-2105-12-208. BMC Bioinformatics. 2011. PMID: 21612673 Free PMC article.
-
A novel side-chain orientation dependent potential derived from random-walk reference state for protein fold selection and structure prediction.PLoS One. 2010 Oct 27;5(10):e15386. doi: 10.1371/journal.pone.0015386. PLoS One. 2010. PMID: 21060880 Free PMC article.
References
-
- Miyazawa S, Jernigan R. Estimation of effective interresidue contact energies from protein crystal structures: quasi-chemical approximation. Macromolecules. 1985;18:534–552. doi: 10.1021/ma00145a039. - DOI
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
