

LIMPIC: a computational method for the separation of protein MALDI-TOF-MS signals from noise
- PMID: 17386085
- PMCID: PMC1847688
- DOI: 10.1186/1471-2105-8-101
LIMPIC: a computational method for the separation of protein MALDI-TOF-MS signals from noise
Abstract
Background: Mass spectrometry protein profiling is a promising tool for biomarker discovery in clinical proteomics. However, the development of a reliable approach for the separation of protein signals from noise is required. In this paper, LIMPIC, a computational method for the detection of protein peaks from linear-mode MALDI-TOF data is proposed. LIMPIC is based on novel techniques for background noise reduction and baseline removal. Peak detection is performed considering the presence of a non-homogeneous noise level in the mass spectrum. A comparison of the peaks collected from multiple spectra is used to classify them on the basis of a detection rate parameter, and hence to separate the protein signals from other disturbances.
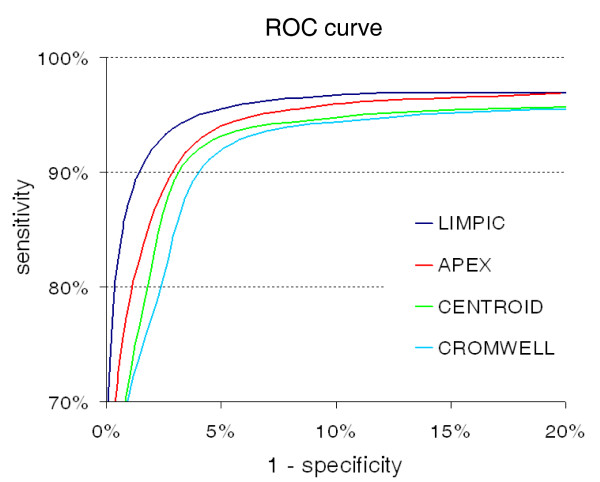
Results: LIMPIC preprocessing proves to be superior than other classical preprocessing techniques, allowing for a reliable decomposition of the background noise and the baseline drift from the MALDI-TOF mass spectra. It provides lower coefficient of variation associated with the peak intensity, improving the reliability of the information that can be extracted from single spectra. Our results show that LIMPIC peak-picking is effective even in low protein concentration regimes. The analytical comparison with commercial and freeware peak-picking algorithms demonstrates its superior performances in terms of sensitivity and specificity, both on in-vitro purified protein samples and human plasma samples.
Conclusion: The quantitative information on the peak intensity extracted with LIMPIC could be used for the recognition of significant protein profiles by means of advanced statistic tools: LIMPIC might be valuable in the perspective of biomarker discovery.
Figures









References
-
- Bonk T, Humeny A. MALDI-TOF-MS analysis of protein and DNA. Neuroscientist. 2001;7:6–12. - PubMed
-
- Maddalo G, Petrucci F, Iezzi M, Pannellini T, Del Boccio P, Ciavardelli D, Biroccio A, Forli F, Di Ilio C, Ballone E, Urbani A, Federici G. Analytical assessment of MALDI-TOF Imaging Mass Spectrometry on thin histological samples. An insight in proteome investigation. Clin Chim Acta. 2005;357:210–218. doi: 10.1016/j.cccn.2005.03.029. - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
