

HMM-ModE--improved classification using profile hidden Markov models by optimising the discrimination threshold and modifying emission probabilities with negative training sequences
- PMID: 17389042
- PMCID: PMC1852395
- DOI: 10.1186/1471-2105-8-104
HMM-ModE--improved classification using profile hidden Markov models by optimising the discrimination threshold and modifying emission probabilities with negative training sequences
Abstract
Background: Profile Hidden Markov Models (HMM) are statistical representations of protein families derived from patterns of sequence conservation in multiple alignments and have been used in identifying remote homologues with considerable success. These conservation patterns arise from fold specific signals, shared across multiple families, and function specific signals unique to the families. The availability of sequences pre-classified according to their function permits the use of negative training sequences to improve the specificity of the HMM, both by optimizing the threshold cutoff and by modifying emission probabilities to minimize the influence of fold-specific signals. A protocol to generate family specific HMMs is described that first constructs a profile HMM from an alignment of the family's sequences and then uses this model to identify sequences belonging to other classes that score above the default threshold (false positives). Ten-fold cross validation is used to optimise the discrimination threshold score for the model. The advent of fast multiple alignment methods enables the use of the profile alignments to align the true and false positive sequences, and the resulting alignments are used to modify the emission probabilities in the original model.
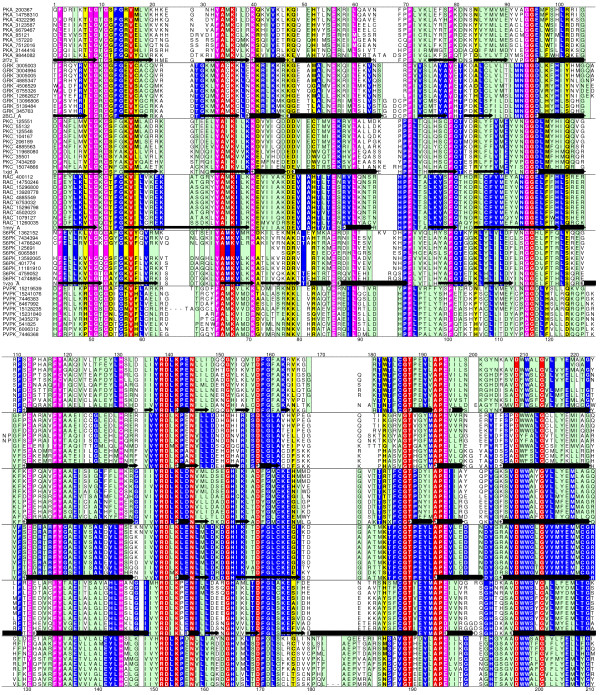
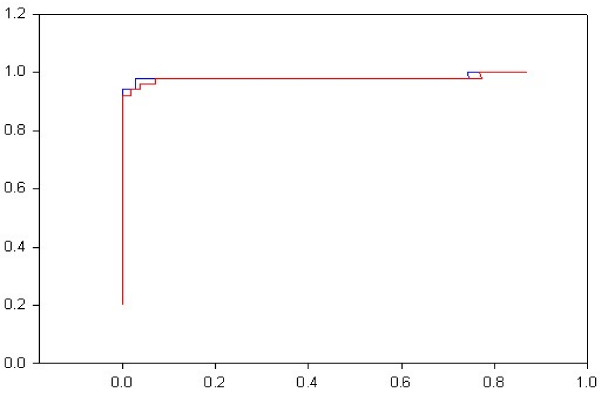
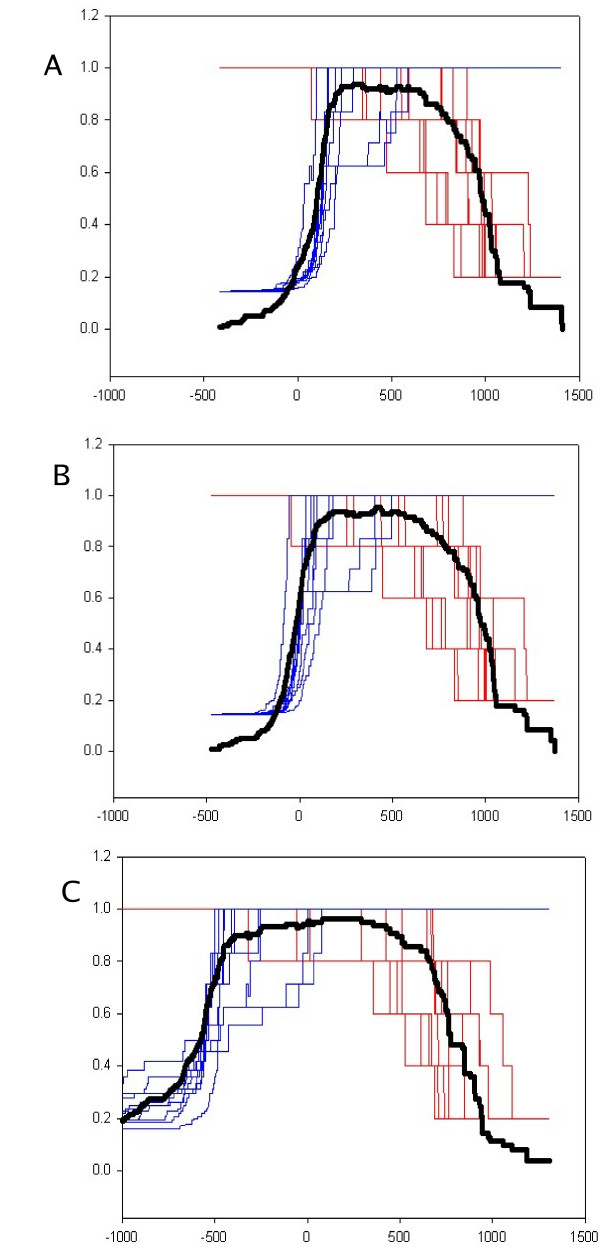
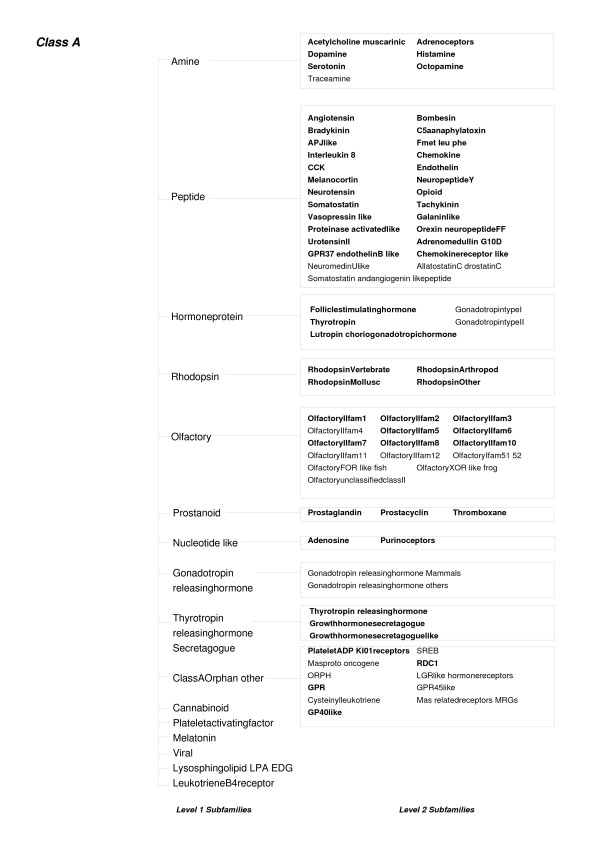
Results: The protocol, called HMM-ModE, was validated on a set of sequences belonging to six sub-families of the AGC family of kinases. These sequences have an average sequence similarity of 63% among the group though each sub-group has a different substrate specificity. The optimisation of discrimination threshold, by using negative sequences scored against the model improves specificity in test cases from an average of 21% to 98%. Further discrimination by the HMM after modifying model probabilities using negative training sequences is provided in a few cases, the average specificity rising to 99%. Similar improvements were obtained with a sample of G-Protein coupled receptors sub-classified with respect to their substrate specificity, though the average sequence identity across the sub-families is just 20.6%. The protocol is applied in a high-throughput classification exercise on protein kinases.
Conclusion: The protocol has the potential to maximise the contributions of discriminating residues to classify proteins based on their molecular function, using pre-classified positive and negative sequence training data. The high specificity of the method, and increasing availability of pre-classified sequence data holds the potential for its application in sequence annotation.
Figures



Similar articles
-
Fast model-based protein homology detection without alignment.Bioinformatics. 2007 Jul 15;23(14):1728-36. doi: 10.1093/bioinformatics/btm247. Epub 2007 May 8. Bioinformatics. 2007. PMID: 17488755
-
Designing patterns for profile HMM search.Bioinformatics. 2007 Jan 15;23(2):e36-43. doi: 10.1093/bioinformatics/btl323. Bioinformatics. 2007. PMID: 17237102
-
Improving profile HMM discrimination by adapting transition probabilities.J Mol Biol. 2004 May 7;338(4):847-54. doi: 10.1016/j.jmb.2004.03.023. J Mol Biol. 2004. PMID: 15099750
-
Sequence comparison and protein structure prediction.Curr Opin Struct Biol. 2006 Jun;16(3):374-84. doi: 10.1016/j.sbi.2006.05.006. Epub 2006 May 19. Curr Opin Struct Biol. 2006. PMID: 16713709 Review.
-
Hidden Markov model and its applications in motif findings.Methods Mol Biol. 2010;620:405-16. doi: 10.1007/978-1-60761-580-4_13. Methods Mol Biol. 2010. PMID: 20652513 Review.
Cited by
-
Rational mutational analysis of a multidrug MFS transporter CaMdr1p of Candida albicans by employing a membrane environment based computational approach.PLoS Comput Biol. 2009 Dec;5(12):e1000624. doi: 10.1371/journal.pcbi.1000624. Epub 2009 Dec 24. PLoS Comput Biol. 2009. PMID: 20041202 Free PMC article.
-
Genomics-driven discovery of a biosynthetic gene cluster required for the synthesis of BII-Rafflesfungin from the fungus Phoma sp. F3723.BMC Genomics. 2019 May 14;20(1):374. doi: 10.1186/s12864-019-5762-6. BMC Genomics. 2019. PMID: 31088369 Free PMC article.
-
Employing information theoretic measures and mutagenesis to identify residues critical for drug-proton antiport function in Mdr1p of Candida albicans.PLoS One. 2010 Jun 10;5(6):e11041. doi: 10.1371/journal.pone.0011041. PLoS One. 2010. PMID: 20548793 Free PMC article.
-
ModEnzA: Accurate Identification of Metabolic Enzymes Using Function Specific Profile HMMs with Optimised Discrimination Threshold and Modified Emission Probabilities.Adv Bioinformatics. 2011;2011:743782. doi: 10.1155/2011/743782. Epub 2011 Mar 29. Adv Bioinformatics. 2011. PMID: 21541071 Free PMC article.
-
Evolutionary history of calcium-sensing receptors unveils hyper/hypocalcemia-causing mutations.PLoS Comput Biol. 2024 Nov 12;20(11):e1012591. doi: 10.1371/journal.pcbi.1012591. eCollection 2024 Nov. PLoS Comput Biol. 2024. PMID: 39531485 Free PMC article.
References
-
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. - PubMed
-
- Eddy SR. HMMER: Profile hidden Markov models for biological sequence analysis. 1998. http://hmmer.janelia.org/
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
