

Animal models for mucopolysaccharidosis disorders and their clinical relevance
- PMID: 17391445
- PMCID: PMC3351033
- DOI: 10.1111/j.1651-2227.2007.00211.x
Animal models for mucopolysaccharidosis disorders and their clinical relevance
Abstract
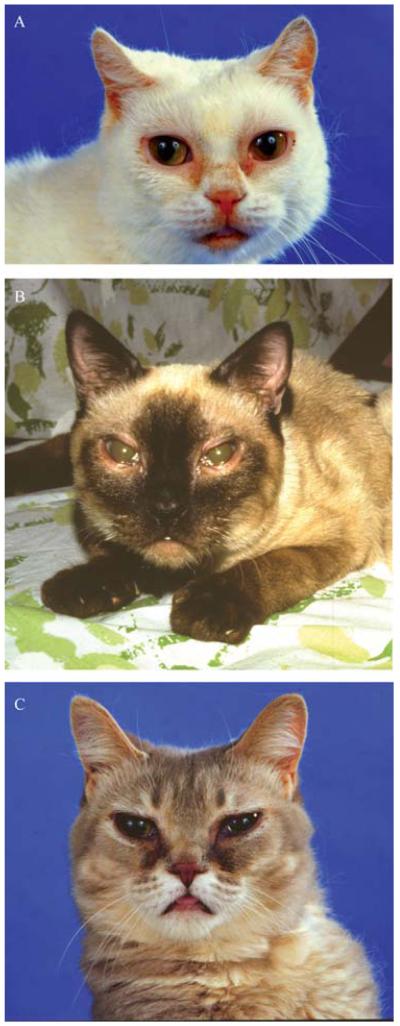
Progress in understanding how a particular genotype produces the phenotype of an inborn error of metabolism, such as a mucopolysaccharidosis, in human patients has been facilitated by the study of animals with mutations in the orthologous genes. These are not just animal models, but true orthologues of the human genetic disease, with defects involving the same evolutionarily conserved genes and the same molecular, biochemical, and anatomic lesions as in human patients. These animals are often domestic species because of the individual medical attention paid to them, particularly dogs and cats. In addition, naturally occurring mouse models have also been found in breeding colonies. Within the last several decades, advances in molecular biology have allowed the production of knockout mouse models of human genetic disease, including the lysosomal storage diseases. The ability to use both inbred strains of a small, prolific species together with larger out-bred animals found because of their disease phenotype provides a powerful combination with which to investigate pathogenesis, develop approaches to therapy, and define biomarkers to evaluate therapeutic success. This has been true for the inborn errors of metabolism and, in particular, the mucopolysaccharidoses.
Conclusion: Animal models of human genetic disease continue to play an important role in understanding the molecular and physiological consequences of lysosomal storage diseases and to provide an opportunity to evaluate the efficacy and safety of therapeutic interventions.
Figures

Comment in
-
Update on mucopolysaccharidosis type II.Acta Paediatr. 2007 Apr;96(455):55. doi: 10.1111/j.1651-2227.2007.00210.x. Acta Paediatr. 2007. PMID: 17391444 No abstract available.
References
-
- Patterson DF, Haskins ME, Jezyk PF, Giger U, Meyers-Wallen VN, Aguirre G, et al. Research on genetic diseases: reciprocal benefits to animals and man. J Am Vet Med Assoc. 1988;193:1131–44. - PubMed
-
- Phaneuf D, Wakamatsu N, Huang JQ, Borowski A, Peterson AC, Fortunato SR, et al. Dramatically different phenotypes in mouse models of human Tay-Sachs and Sandhoff diseases. Hum Mol Genet. 1996;5:1–14. - PubMed
-
- Cohen-Tannoudji M, Marchand P, Akli S, Sheardown SA, Puech JP, Kress C, et al. Disruption of murine Hexa gene leadsto enzymatic deficiency and to neuronal lysosomal storage, similar to that observed in Tay-Sachs disease. Mamm Genome. 1995;6:844–9. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
