

Autism, language delay and mental retardation in a patient with 7q11 duplication
- PMID: 17400790
- PMCID: PMC1994965
- DOI: 10.1136/jmg.2006.047092
Autism, language delay and mental retardation in a patient with 7q11 duplication
Abstract
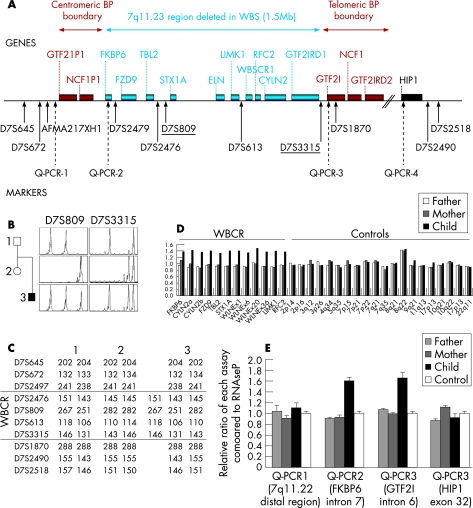
Background: Chromosomal rearrangements, arising from unequal recombination between repeated sequences, are found in a subset of patients with autism. Duplications involving loci associated with behavioural disturbances constitute an especially good candidate mechanism. The Williams-Beuren critical region (WBCR), located at 7q11.23, is commonly deleted in Williams-Beuren microdeletion syndrome (WBS). However, only four patients with a duplication of the WBCR have been reported to date: one with severe language delay and the three others with variable developmental, psychomotor and language delay.
Objective and methods: In this study, we screened 206 patients with autism spectrum disorders for the WBCR duplication by quantitative microsatellite analysis and multiple ligation-dependent probe amplification.
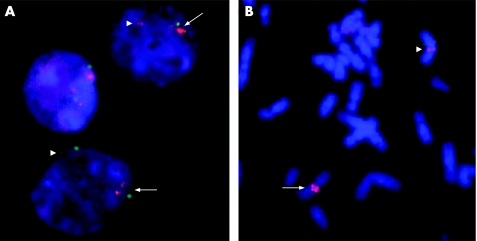
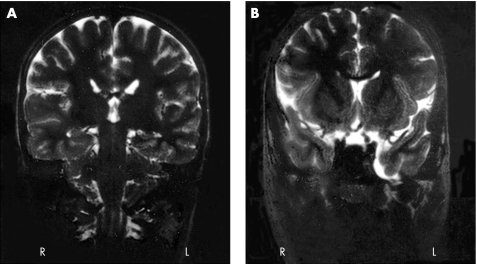
Results: We identified one male patient with a de novo interstitial duplication of the entire WBCR of paternal origin. The patient had autistic disorder, severe language delay and mental retardation, with very mild dysmorphic features.
Conclusion: We report the first patient with autistic disorder and a WBCR duplication. This observation indicates that the 7q11.23 duplication could be involved in complex clinical phenotypes, ranging from developmental or language delay to mental retardation and autism, and extends the phenotype initially reported. These findings also support the existence of one or several genes in 7q11.23 sensitive to gene dosage and involved in the development of language and social interaction.
Conflict of interest statement
Competing interests: None declared.
Comment in
-
Duplication of the Williams-Beuren critical region: case report and further delineation of the phenotypic spectrum.J Med Genet. 2008 Mar;45(3):187-9. doi: 10.1136/jmg.2007.054064. J Med Genet. 2008. PMID: 18310268 No abstract available.
References
-
- Thomas N S, Durkie M, Potts G, Sandford R, Van Zyl B, Youings S, Dennis N R, Jacobs P A. Parental and chromosomal origins of microdeletion and duplication syndromes involving 7q11.23, 15q11‐q13 and 22q11. Eur J Hum Genet 200614831–837. - PubMed
-
- Potocki L, Chen K S, Park S S, Osterholm D E, Withers M A, Kimonis V, Summers A M, Meschino W S, Anyane‐Yeboa K, Kashork C D, Shaffer L G, Lupski J R. Molecular mechanism for duplication 17p11.2 – the homologous recombination reciprocal of the Smith‐Magenis microdeletion. Nat Genet 20002484–87. - PubMed
-
- Baumer A, Dutly F, Balmer D, Riegel M, Tukel T, Krajewska‐Walasek M, Schinzel A A. High level of unequal meiotic crossovers at the origin of the 22q11. 2 and 7q11.23 deletions. Hum Mol Genet 19987887–894. - PubMed
-
- Stromme P, Bjornstad P G, Ramstad K. Prevalence estimation of Williams syndrome. J Child Neurol 200217269–271. - PubMed
-
- Meyer‐Lindenberg A, Mervis C B, Faith Berman K. Neural mechanisms in Williams syndrome: a unique window to genetic influences on cognition and behaviour. Nat Rev Neurosci 20067380–393. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources