

Identification of functional modules using network topology and high-throughput data
- PMID: 17408515
- PMCID: PMC1839897
- DOI: 10.1186/1752-0509-1-8
Identification of functional modules using network topology and high-throughput data
Abstract
Background: With the advent of systems biology, biological knowledge is often represented today by networks. These include regulatory and metabolic networks, protein-protein interaction networks, and many others. At the same time, high-throughput genomics and proteomics techniques generate very large data sets, which require sophisticated computational analysis. Usually, separate and different analysis methodologies are applied to each of the two data types. An integrated investigation of network and high-throughput information together can improve the quality of the analysis by accounting simultaneously for topological network properties alongside intrinsic features of the high-throughput data.
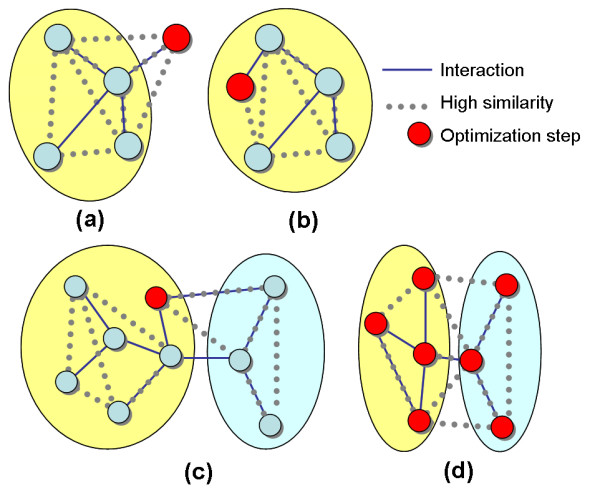
Results: We describe a novel algorithmic framework for this challenge. We first transform the high-throughput data into similarity values, (e.g., by computing pairwise similarity of gene expression patterns from microarray data). Then, given a network of genes or proteins and similarity values between some of them, we seek connected sub-networks (or modules) that manifest high similarity. We develop algorithms for this problem and evaluate their performance on the osmotic shock response network in S. cerevisiae and on the human cell cycle network. We demonstrate that focused, biologically meaningful and relevant functional modules are obtained. In comparison with extant algorithms, our approach has higher sensitivity and higher specificity.
Conclusion: We have demonstrated that our method can accurately identify functional modules. Hence, it carries the promise to be highly useful in analysis of high throughput data.
Figures








References
-
- Lord P, Stevens R, Brass A, Goble C. Investigating semantic similarity measures across the Gene Ontology: the relationship between sequence and annotation. Bioinformatics. 2003;19:1275–83. - PubMed
-
- Ge H, Liu Z, Church G, Vidal M. Correlation between transcriptome and interactome mapping data from Saccharomyces cerevisiae. Nat Genet. 2001;29:482–486. - PubMed
-
- Jansen R, Yu H, Greenbaum D, Kluger Y, Krogan NJ, Chung S, Emili A, Snyder M, Greenblatt JF, Gerstein M. A Bayesian networks approach for predicting protein-protein interactions from genomic data. Science. 2003;302 - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
