

An exact nonparametric method for inferring mosaic structure in sequence triplets
- PMID: 17409078
- PMCID: PMC1894573
- DOI: 10.1534/genetics.106.068874
An exact nonparametric method for inferring mosaic structure in sequence triplets
Abstract
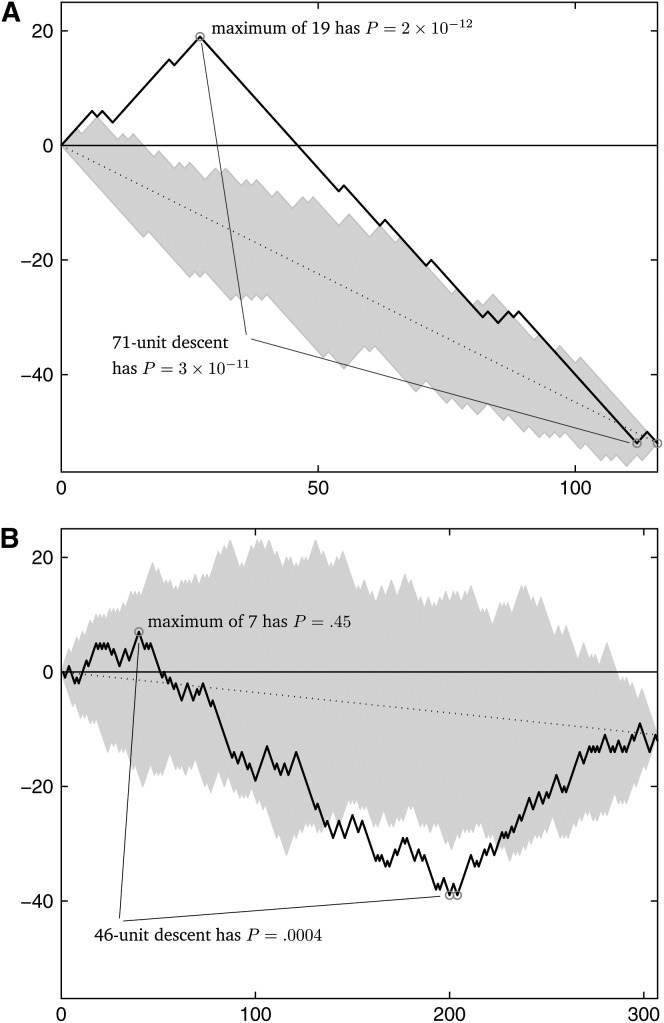
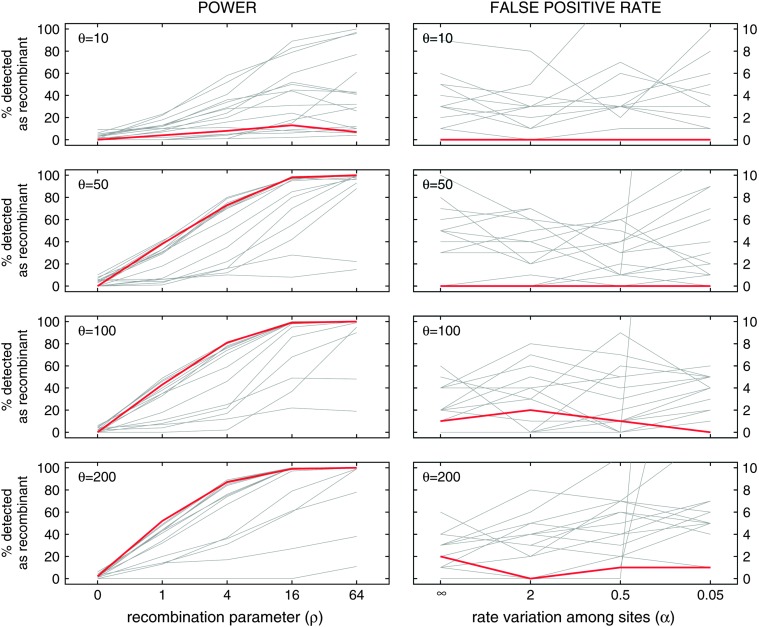
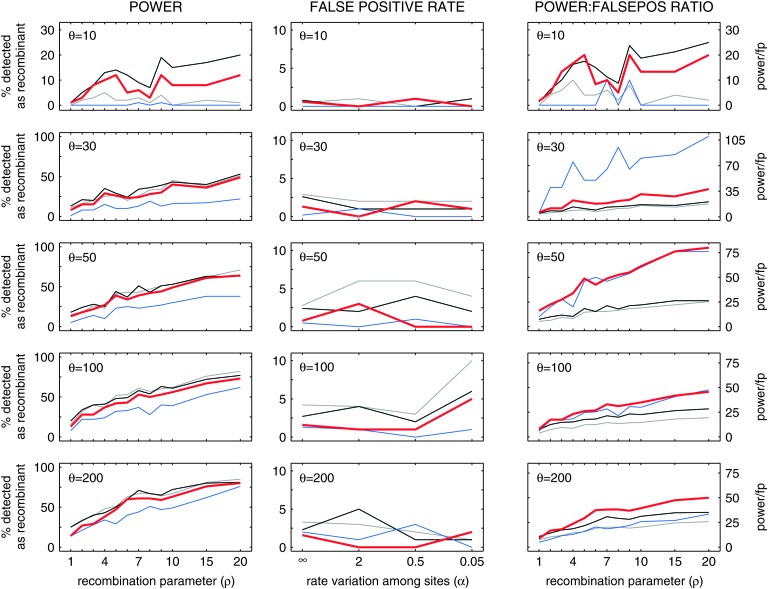
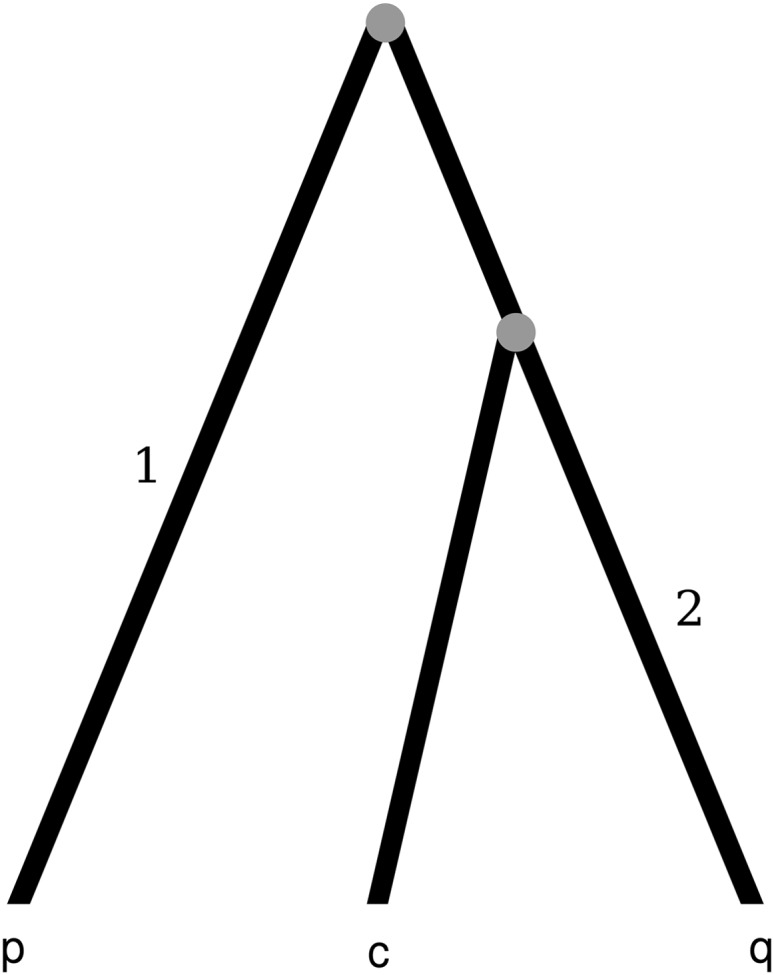
Statistical tests for detecting mosaic structure or recombination among nucleotide sequences usually rely on identifying a pattern or a signal that would be unlikely to appear under clonal reproduction. Dozens of such tests have been described, but many are hampered by long running times, confounding of selection and recombination, and/or inability to isolate the mosaic-producing event. We introduce a test that is exact, nonparametric, rapidly computable, free of the infinite-sites assumption, able to distinguish between recombination and variation in mutation/fixation rates, and able to identify the breakpoints and sequences involved in the mosaic-producing event. Our test considers three sequences at a time: two parent sequences that may have recombined, with one or two breakpoints, to form the third sequence (the child sequence). Excess similarity of the child sequence to a candidate recombinant of the parents is a sign of recombination; we take the maximum value of this excess similarity as our test statistic Delta(m,n,b). We present a method for rapidly calculating the distribution of Delta(m,n,b) and demonstrate that it has comparable power to and a much improved running time over previous methods, especially in detecting recombination in large data sets.
Figures
References
-
- Ardlie, K. G., L. Kruglyak and M. Seielstad, 2002. Patterns of linkage disequilbrium in the human genome. Nat. Rev. Genet. 3 299–309. - PubMed
-
- Awadalla, P., 2003. The evolutionary genomics of pathogen recombination. Nat. Rev. Genet. 4 50–60. - PubMed
-
- Awadalla, P., A. Eyre-Walker and J. Maynard Smith, 1999. Linkage disequilibrium and recombination in hominid mitochondrial DNA. Science 286 2524–2525. - PubMed
-
- Balding, D. J., R. A. Nichols and D. M. Hunt, 1992. Detecting gene conversion: primate visual pigment genes. Proc. R. Soc. Lond. Ser. B 249 275–280. - PubMed
-
- Barton, D. E., and C. L. Mallows, 1965. Some aspects of the random sequence. Ann. Math. Stat. 36 236–260.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
