

Efficient target-selected mutagenesis in Caenorhabditis elegans: toward a knockout for every gene
- PMID: 17416746
- PMCID: PMC1855173
- DOI: 10.1101/gr.6080607
Efficient target-selected mutagenesis in Caenorhabditis elegans: toward a knockout for every gene
Abstract
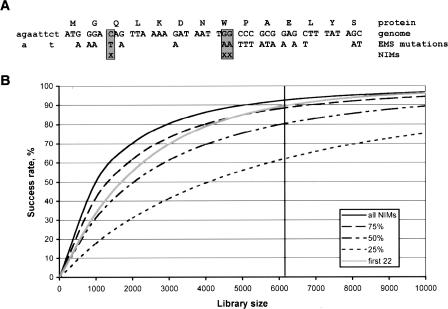
Reverse genetic or gene-driven knockout approaches have contributed significantly to the success of model organisms for fundamental and biomedical research. Although various technologies are available for C. elegans, none of them scale very well for genome-wide application. To address this, we implemented a target-selected knockout approach that is based on random chemical mutagenesis and detection of single nucleotide mutations in genes of interest using high-throughput resequencing. A clonal library of 6144 EMS-mutagenized worms was established and screened, resulting in the identification of 1044 induced mutations in 109 Mbp, which translates into an average spacing between exonic mutations in the library of only 17 bp. We covered 25% of the open reading frames of 32 genes and identified one or more inactivating mutations (nonsense or splice site) in 84% of them. Extrapolation of our results indicates that nonsense mutations for >90% of all C. elegans genes are present in the library. To identify all of these mutations, one only needs to inspect those positions that--given the known specificity of the mutagen--can result in the introduction of a stop codon. We define these positions as nonsense introducing mutations (NIMs). The genome-wide collection of possible NIMs can be calculated for any organism with a sequenced genome and reduces the screening complexity by 200- to 2000-fold, depending on the organism and mutagen. For EMS-mutagenized C. elegans, there are only approximately 500,000 NIMs. We show that a NIM genotyping approach employing high-density microarrays can, in principle, be used for the genome-wide identification of C. elegans knockouts.
Figures



References
-
- Anderson P. Mutagenesis. Methods Cell Biol. 1995;48:31–58. - PubMed
-
- Barstead R.J. Reverse genetics. In: Hope I.A., editor. C. elegans: —A Practical Approach. Oxford University Press; New York: 1999. pp. 97–118.
MeSH terms
LinkOut - more resources
Full Text Sources
Miscellaneous