

The mode and tempo of genome size evolution in eukaryotes
- PMID: 17420184
- PMCID: PMC1855170
- DOI: 10.1101/gr.6096207
The mode and tempo of genome size evolution in eukaryotes
Erratum in
- Genome Res. 2010 Jun;20(6):874
Abstract
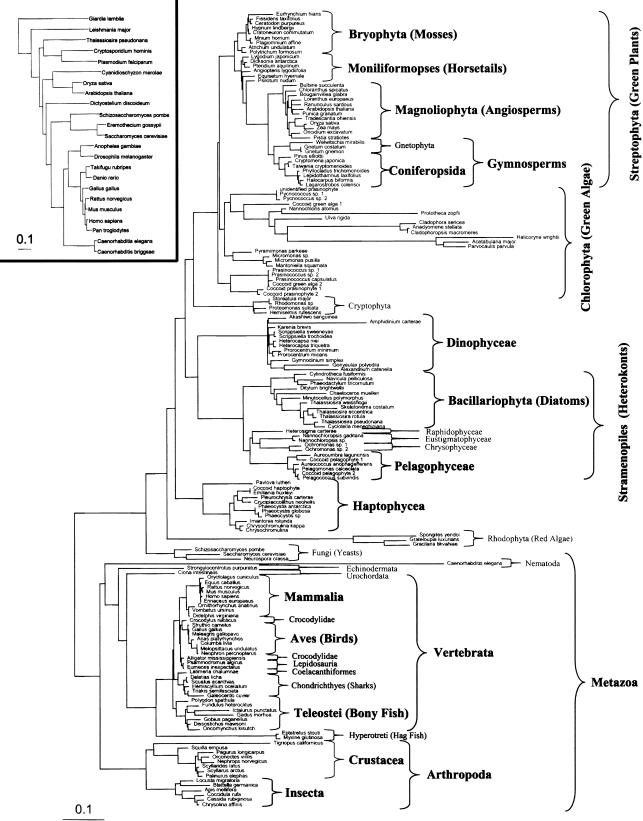
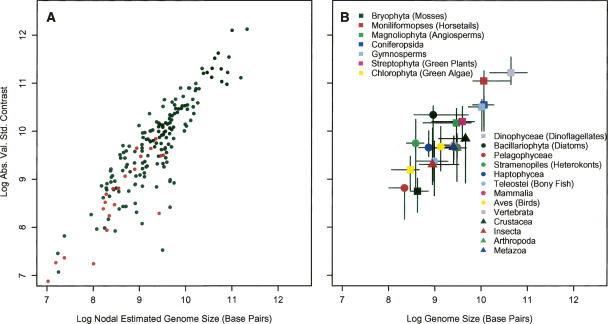
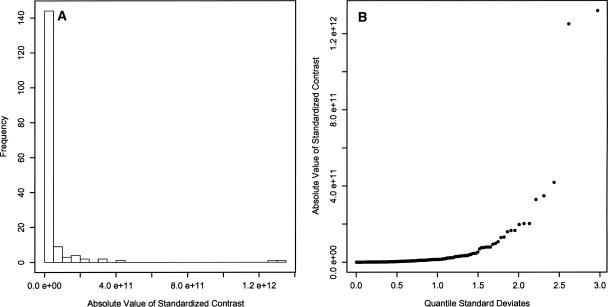
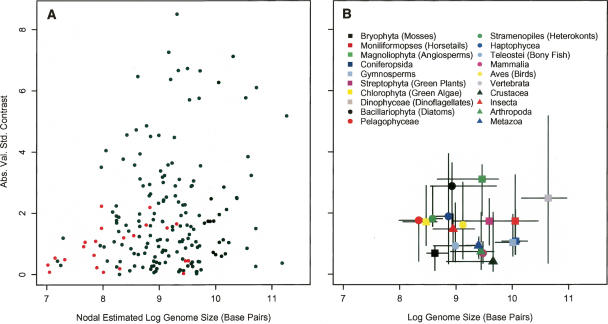
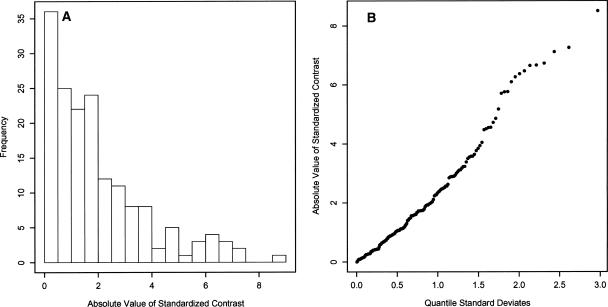
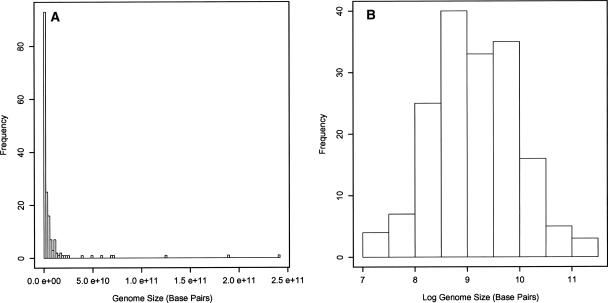
Eukaryotic genome size varies over five orders of magnitude; however, the distribution is strongly skewed toward small values. Genome size is highly correlated to a number of phenotypic traits, suggesting that the relative lack of large genomes in eukaryotes is due to selective removal. Using phylogenetic contrasts, we show that the rate of genome size evolution is proportional to genome size, with the fastest rates occurring in the largest genomes. This trend is evident across the 20 major eukaryotic clades analyzed, indicating that over long time scales, proportional change is the dominant and universal mode of genome-size evolution in eukaryotes. Our results reveal that the evolution of eukaryotic genome size can be described by a simple proportional model of evolution. This model explains the skewed distribution of eukaryotic genome sizes without invoking strong selection against large genomes.
Figures
References
-
- Albertini A.M., Hofer M., Calos M.P., Miller J.H., Hofer M., Calos M.P., Miller J.H., Calos M.P., Miller J.H., Miller J.H. On the formation of spontaneous deletions: The importance of short sequence homologies in the generation of large deletions. Cell. 1982;29:319–328. - PubMed
-
- Bookstein F.L. Random walk and the existence of evolutionary rates. Paleobiology. 1987;13:446–464.
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources