

CDK4 and CDK6 delay senescence by kinase-dependent and p16INK4a-independent mechanisms
- PMID: 17420273
- PMCID: PMC1900050
- DOI: 10.1128/MCB.02286-06
CDK4 and CDK6 delay senescence by kinase-dependent and p16INK4a-independent mechanisms
Abstract
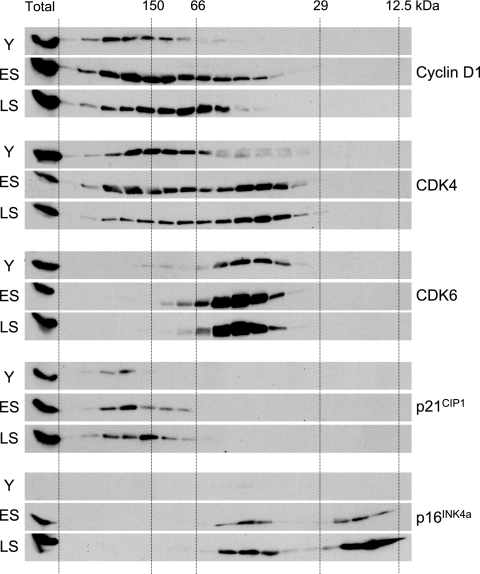
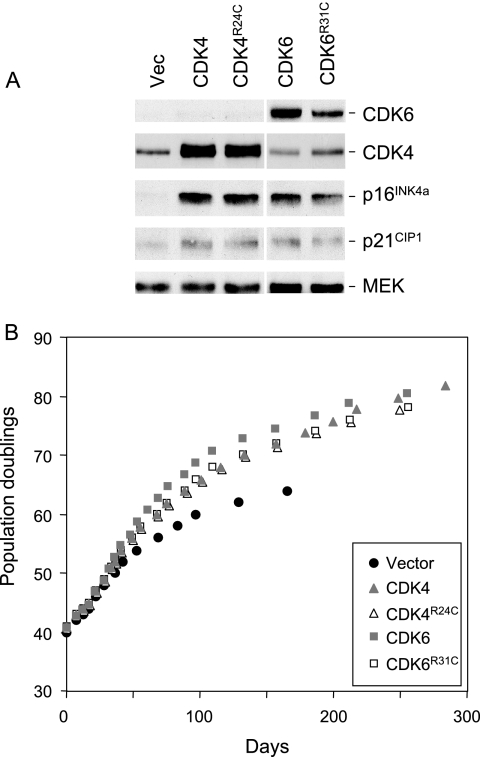
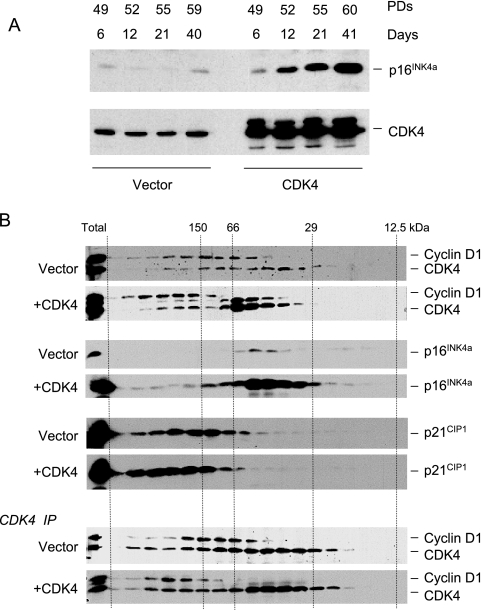
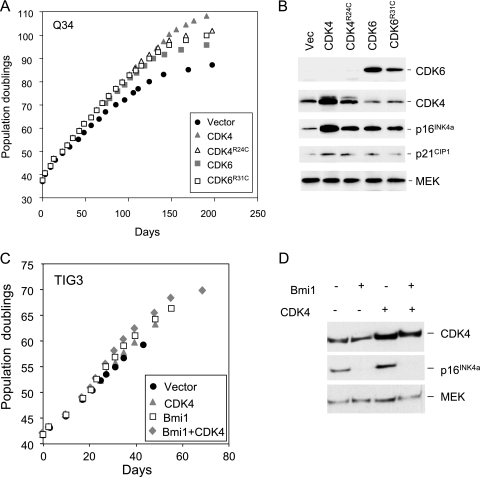
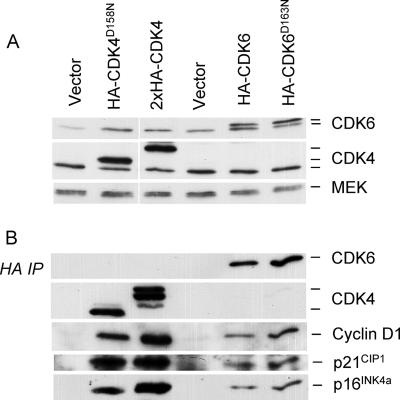
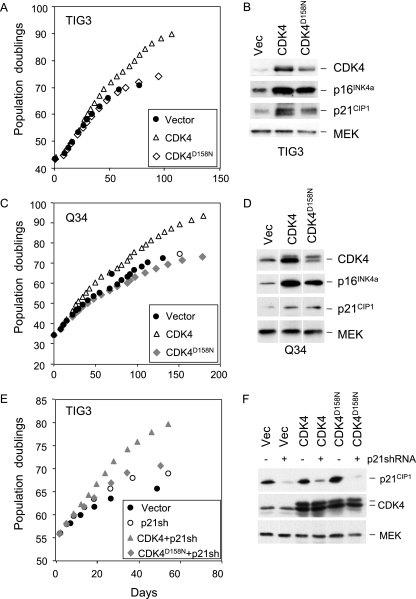
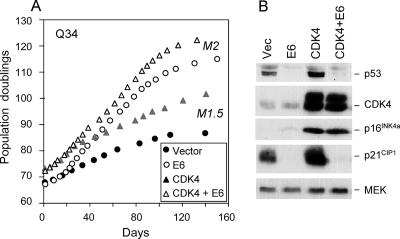
Replicative senescence of human diploid fibroblasts (HDFs) is largely implemented by the cyclin-dependent kinase (CDK) inhibitors p16(INK4a) and p21(CIP1). Their accumulation results in a loss of CDK2 activity, and cells arrest with the retinoblastoma protein (pRb) in its hypophosphorylated state. It has become standard practice to bypass the effects of p16(INK4a) by overexpressing CDK4 or a variant form that is unable to bind to INK4 proteins. Although CDK4 and CDK6 and their INK4-insensitive variants can extend the life span of HDFs, they also cause a substantial increase in the levels of endogenous p16(INK4a). Here we show that CDK4 and CDK6 can extend the life span of HDFs that have inactivating mutations in both alleles of INK4a or in which INK4a levels are repressed, indicating that overexpression of CDK4/6 is not equivalent to ablation of p16(INK4a). However, catalytically inactive versions of these kinases are unable to extend the replicative life span, suggesting that the impact of ectopic CDK4/6 depends on their ability to phosphorylate as yet unidentified substrates rather than to sequester CDK inhibitors. Since p16(INK4a) deficiency, CDK4 expression, and p53 or p21(CIP1) ablation have additive effects on replicative life span, our results underscore the idea that senescence is an integrated response to diverse signals.
Figures
References
-
- Baker, G. L., M. W. Landis, and P. W. Hinds. 2005. Multiple functions of D-type cyclins can antagonize pRb-mediated suppression of proliferation. Cell Cycle 4:330-338. - PubMed
-
- Bates, S., L. Bonetta, D. MacAllan, D. Parry, A. Holder, C. Dickson, and G. Peters. 1994. CDK6 (PLSTIRE) and CDK4 (PSK-J3) are a distinct subset of the cyclin-dependent kinases that associate with cyclin D1. Oncogene 9:71-79. - PubMed
-
- Berthet, C., E. Aleem, V. Coppola, L. Tessarollo, and P. Kaldis. 2003. Cdk2 knockout mice are viable. Curr. Biol. 13:1775-1785. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous