

Peroxisome proliferator-activated receptor-gamma agonists inhibit respiratory syncytial virus-induced expression of intercellular adhesion molecule-1 in human lung epithelial cells
- PMID: 17425601
- PMCID: PMC2265928
- DOI: 10.1111/j.1365-2567.2006.02539.x
Peroxisome proliferator-activated receptor-gamma agonists inhibit respiratory syncytial virus-induced expression of intercellular adhesion molecule-1 in human lung epithelial cells
Abstract
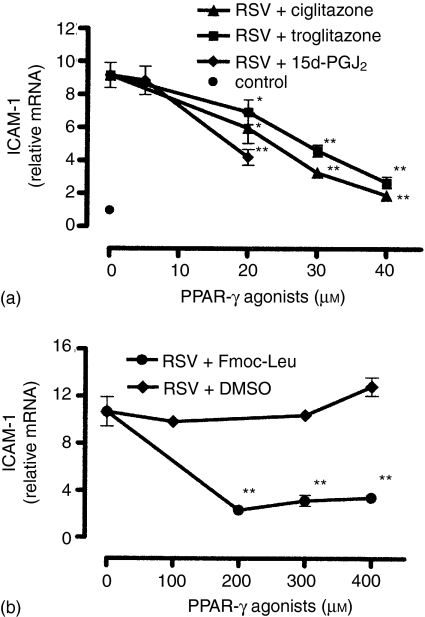
Respiratory syncytial virus (RSV) is the major causative agent of severe lower respiratory tract disease and death in infants worldwide. The epithelial cells of the airways are the target cells for RSV infection and the site of the majority of the inflammation associated with the disease. However, despite five decades of intensive RSV research there exist neither an effective active vaccine nor a promising antiviral and anti-inflammatory therapy. Recently, peroxisome proliferator-activated receptor-gamma (PPAR-gamma), a member of the nuclear hormone receptor superfamily, has been shown to possess anti-inflammatory properties. Therefore, we hypothesized whether the detrimental increase of intercellular adhesion molecule-1 (ICAM-1) on RSV-infected lung epithelial cells (A549 and primary normal human bronchial epithelial cells (NHBE)) might be modulated by natural and synthetic PPAR-gamma agonists (15d-PGJ2, ciglitazone, troglitazone, Fmoc-Leu). Our data show that all PPAR-gamma agonists under study significantly down-regulated the RSV-induced expression of ICAM-1 on A549- and NHBE cells in a dose-dependent manner resulting in a reduced beta2 integrin-mediated adhesion of monocytic effector cells (U937) to RSV-infected A549 cell monolayers. In contrast, the PPAR-alpha agonist bezafibrate had no impact on the RSV-induced ICAM-1 expression. The reduced ICAM-1 expression was associated with a diminished ICAM-1 mRNA level and binding activity of nuclear factor-kappaB (p65/p50) in A549 cells. These findings suggest that PPARgamma agonists have beneficial effects in the suppression of the inflammatory response during RSV infection and therefore might have clinical efficacy in the course of severe RSV-infection.
Figures






References
-
- Hall CB, McCarthy CA. Respiratory syncytial virus. In: Mandell GL, Benett JE, Dolin R, editors. Principles and Practice of Infectious Diseases. Philadelphia: Churchill Livingstone; 2000. pp. 1782–801.
-
- Kimpen JLL. Management of respiratory syncytial virus infection. Curr Opin Infect Dis. 2001;14:323–8. - PubMed
-
- Kneyber MCJ, Moll HA, de Groot R. Treatment and prevention of respiratory syncytial virus infection. Eur J Pediatr. 2000;159:399–411. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
