

Tissue-specific mechanisms for CCN2/CTGF persistence in fibrotic gingiva: interactions between cAMP and MAPK signaling pathways, and prostaglandin E2-EP3 receptor mediated activation of the c-JUN N-terminal kinase
- PMID: 17428796
- PMCID: PMC2443949
- DOI: 10.1074/jbc.M610432200
Tissue-specific mechanisms for CCN2/CTGF persistence in fibrotic gingiva: interactions between cAMP and MAPK signaling pathways, and prostaglandin E2-EP3 receptor mediated activation of the c-JUN N-terminal kinase
Abstract
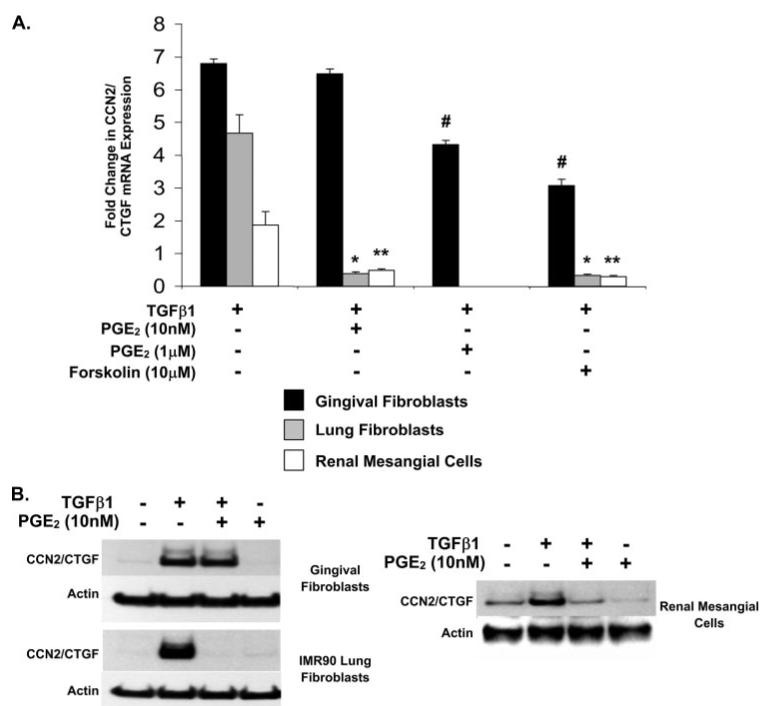
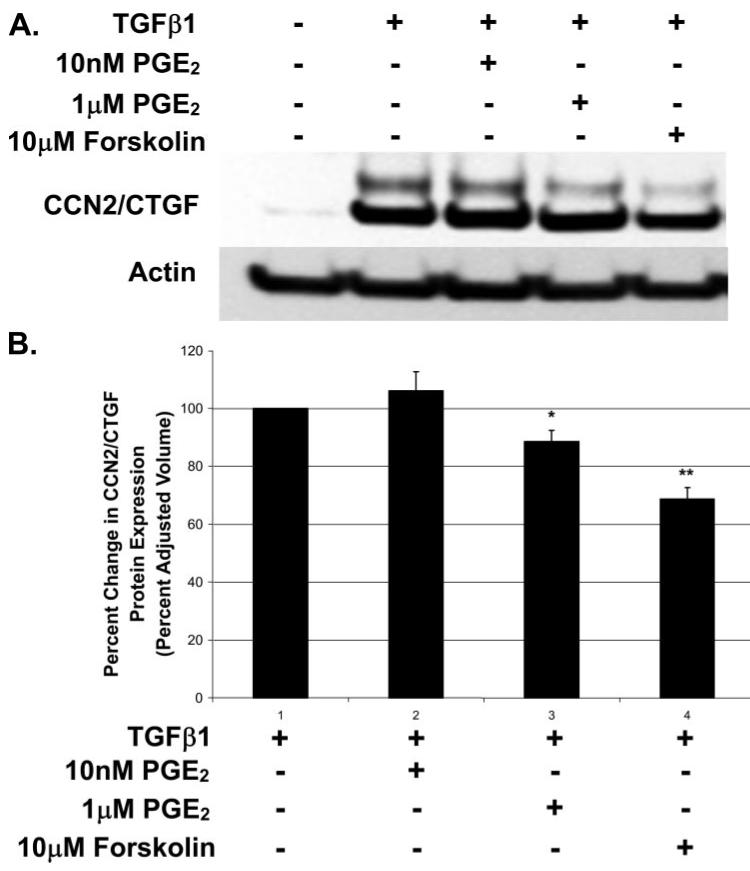
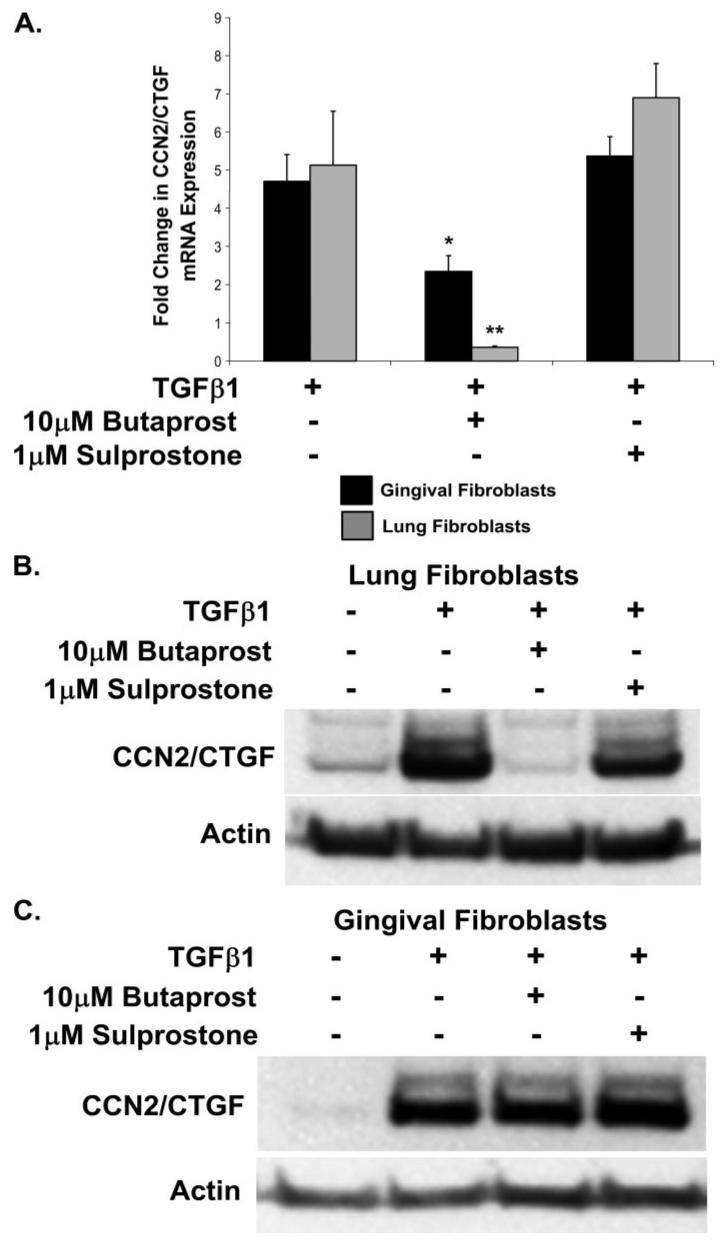
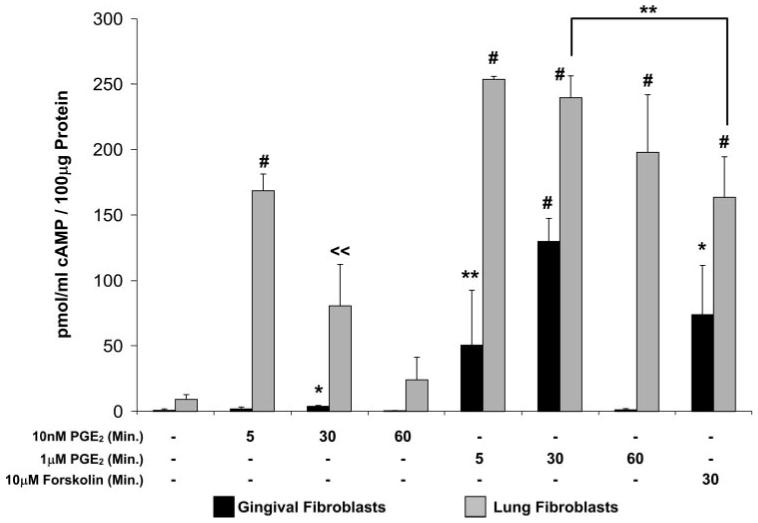
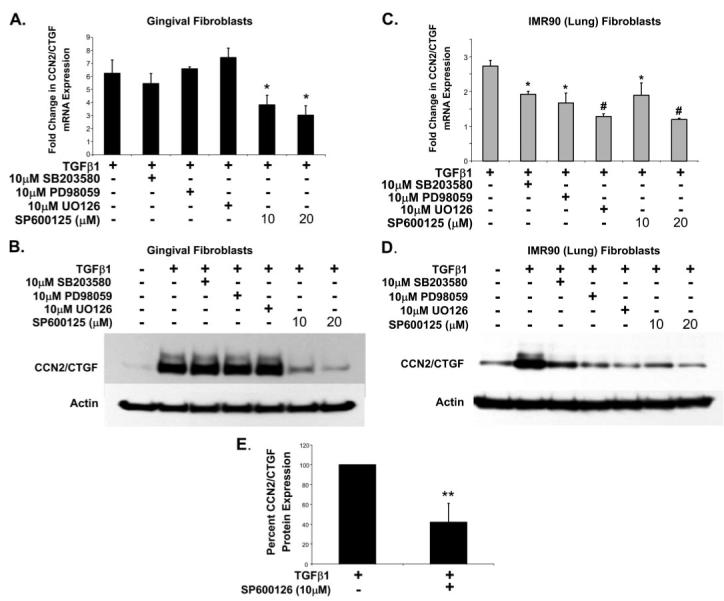
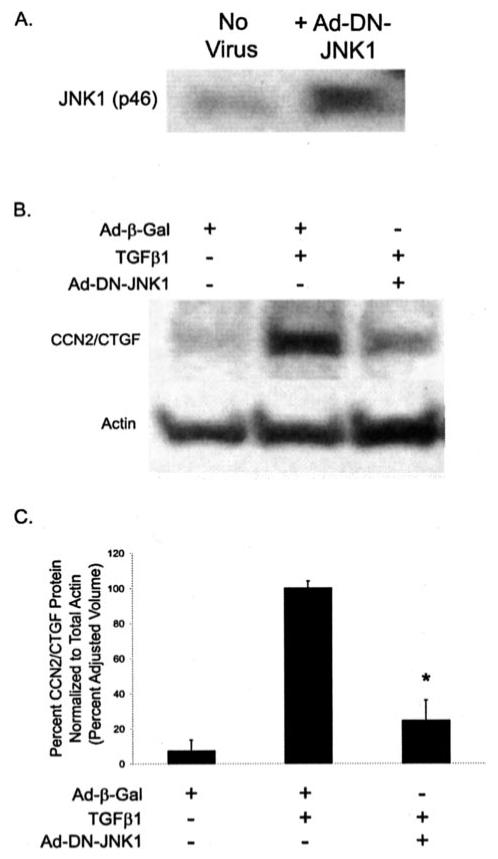
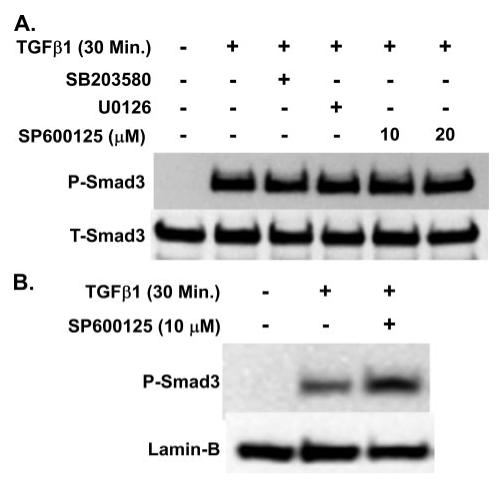
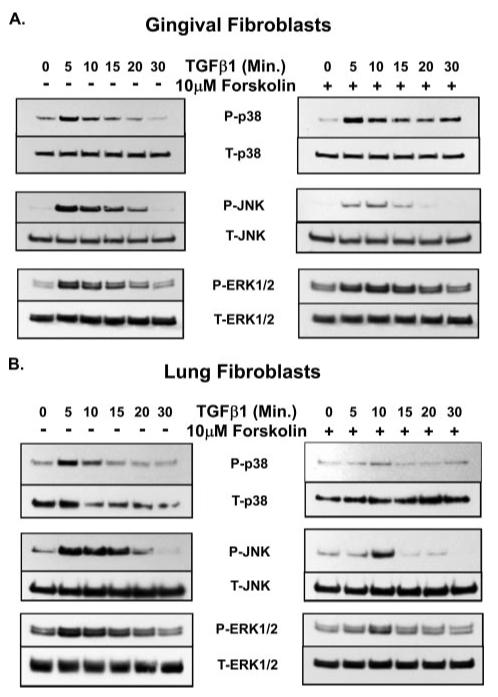
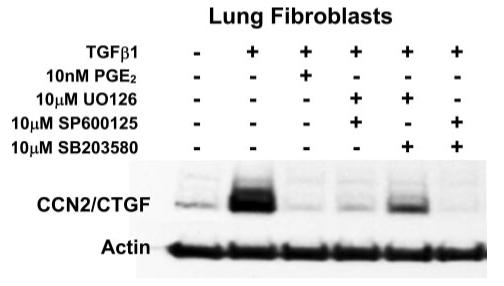
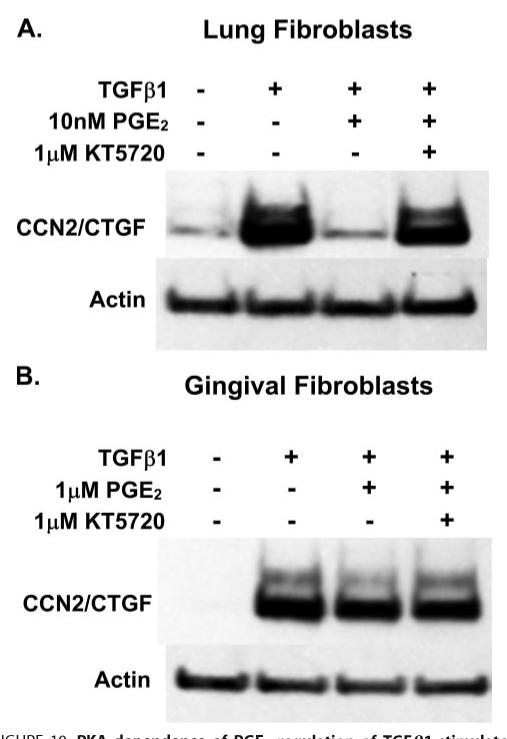
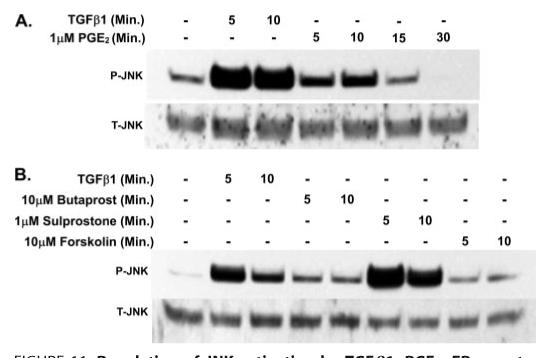
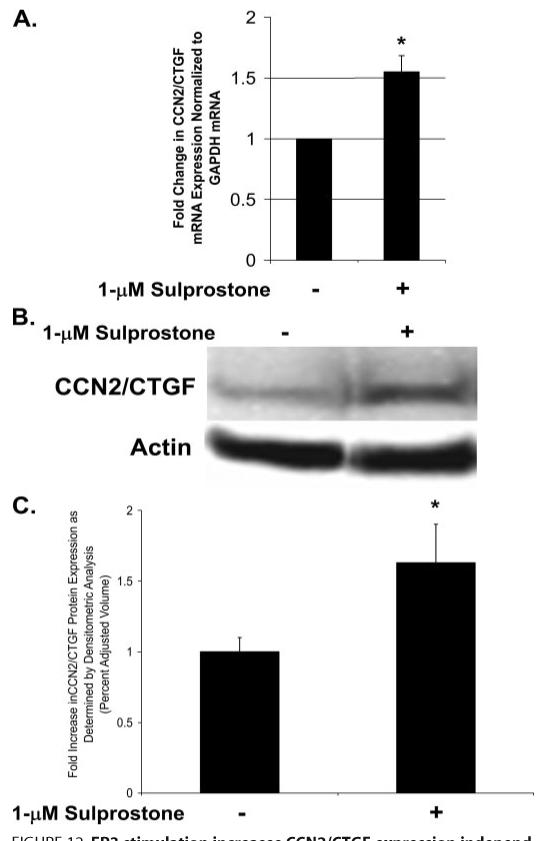
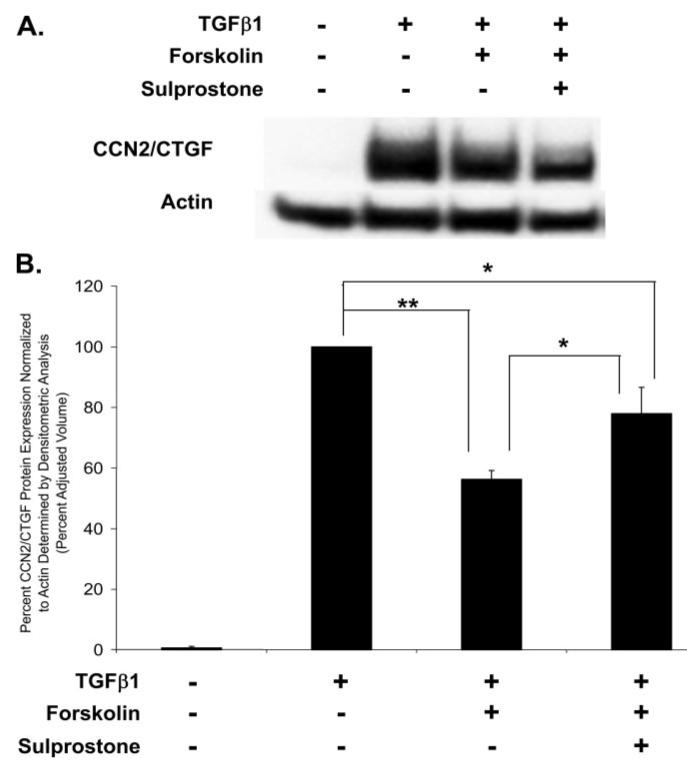
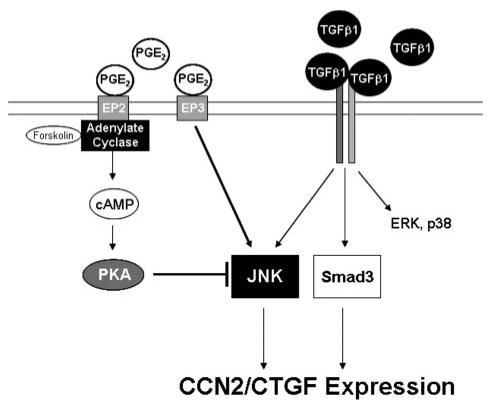
Prostaglandin E(2) blocks transforming growth factor TGF beta1-induced CCN2/CTGF expression in lung and kidney fibroblasts. PGE(2) levels are high in gingival tissues yet CCN2/CTGF expression is elevated in fibrotic gingival overgrowth. Gingival fibroblast expression of CCN2/CTGF in the presence of PGE(2) led us to compare the regulation of CCN2/CTGF expression in fibroblasts cultured from different tissues. Data demonstrate that the TGFbeta1-induced expression of CCN2/CTGF in human lung and renal mesangial cells is inhibited by 10 nm PGE(2), whereas human gingival fibroblasts are resistant. Ten nm PGE(2) increases cAMP accumulation in lung but not gingival fibroblasts, which require 1 mum PGE(2) to elevate cAMP. Micromolar PGE(2) only slightly reduces the TGFbeta1-stimulated CCN2/CTGF levels in gingival cells. EP2 prostaglandin receptor activation with butaprost blocks the TGFbeta1-stimulated expression of CCN2/CTGF expression in lung, but not gingival, fibroblasts. In lung fibroblasts, inhibition of the TGFbeta1-stimulated CCN2/CTGF by PGE(2), butaprost, or forskolin is due to p38, ERK, and JNK MAP kinase inhibition that is cAMP-dependent. Inhibition of any two MAPKs completely blocks CCN2/CTGF expression stimulated by TGFbeta1. These data mimic the inhibitory effects of 10 nm PGE(2) and forskolin that were dependent on PKA activity. In gingival fibroblasts, the sole MAPK mediating the TGFbeta1-stimulated CCN2/CTGF expression is JNK. Whereas forskolin reduces TGFbeta1-stimulated expression of CCN2/CTGF by 35% and JNK activation in gingival fibroblasts, micromolar PGE(2)-stimulated JNK in gingival fibroblasts and opposes the inhibitory effects of cAMP on CCN2/CTGF expression. Stimulation of the EP3 receptor with sulprostone results in a robust increase in JNK activation in these cells. Taken together, data identify two mechanisms by which TGFbeta1-stimulated CCN2/CTGF levels in human gingival fibroblasts resist down-regulation by PGE(2): (i) cAMP cross-talk with MAPK pathways is limited in gingival fibroblasts; (ii) PGE(2) activation of the EP3 prostanoid receptor stimulates the activation of JNK.
Figures
References
-
- Oemar BS, Luscher TF. Arterioscler. Thromb. Vasc. Biol. 1997;17:1483–1489. - PubMed
-
- Brigstock DR. Endocr. Rev. 1999;20:189–206. - PubMed
-
- Bork P. FEBS Lett. 1993;327:125–130. - PubMed
-
- Moussad EE, Brigstock DR. Mol. Genet. Metab. 2000;71:276–292. - PubMed
-
- Chujo S, Shirasaki F, Kawara S, Inagaki Y, Kinbara T, Inaoki M, Takigawa M, Takehara K. J. Cell. Physiol. 2005;203:447–456. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
