

Estimating the number of ancestral lineages using a maximum-likelihood method based on rejection sampling
- PMID: 17435232
- PMCID: PMC1931561
- DOI: 10.1534/genetics.106.066233
Estimating the number of ancestral lineages using a maximum-likelihood method based on rejection sampling
Abstract
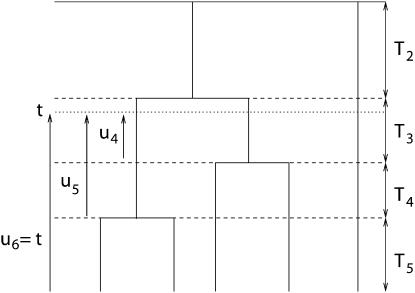
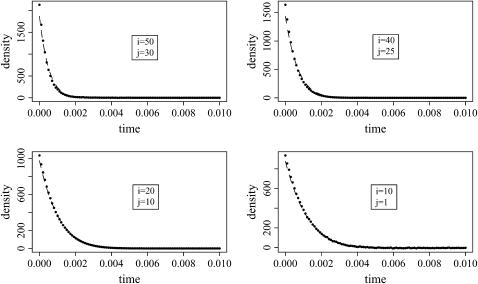
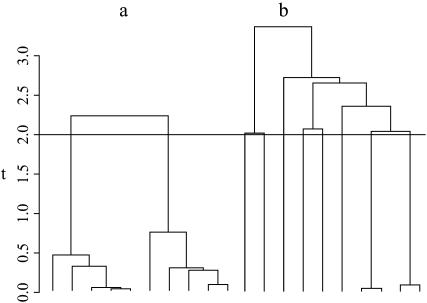
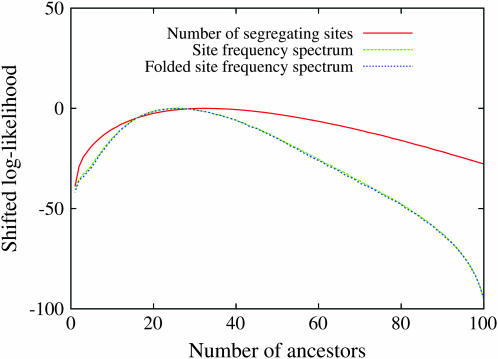
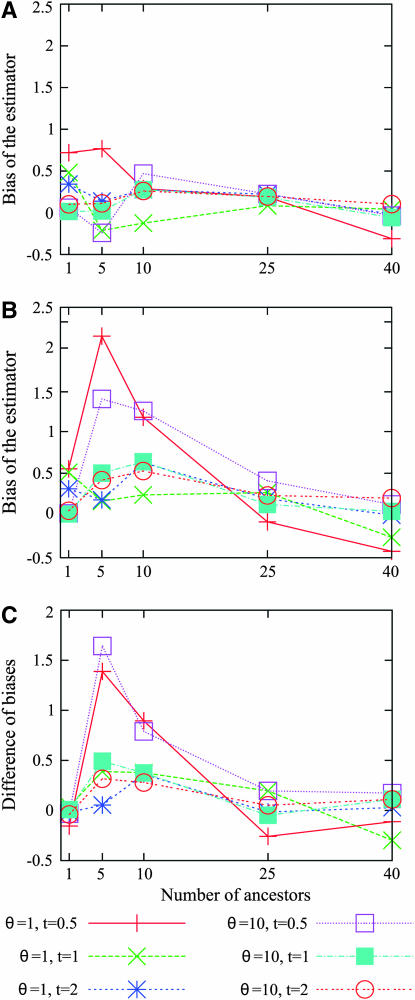
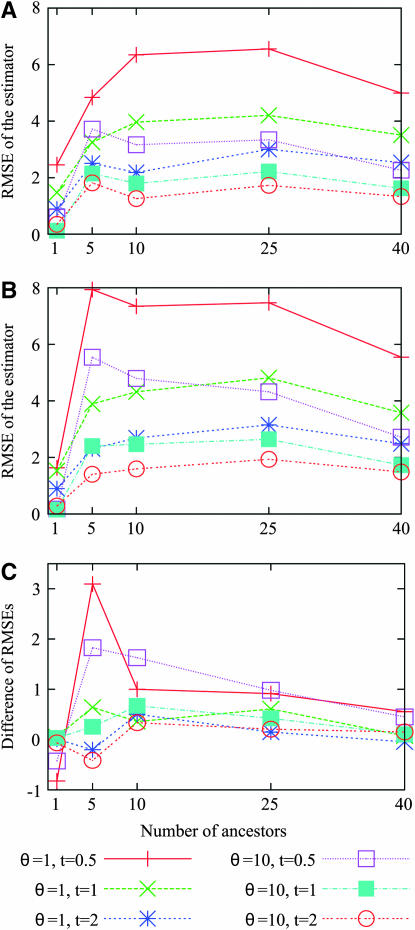
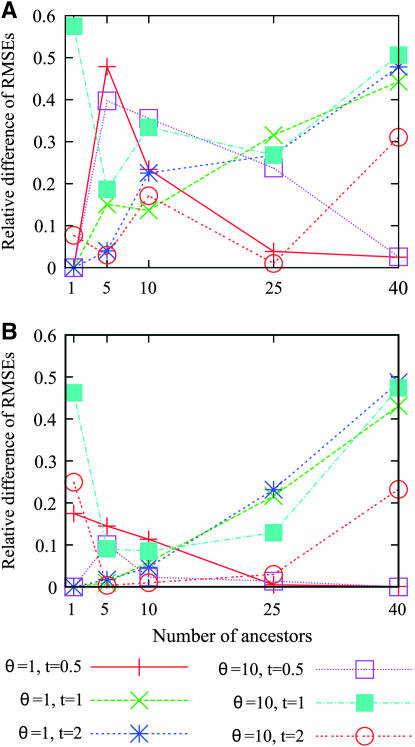
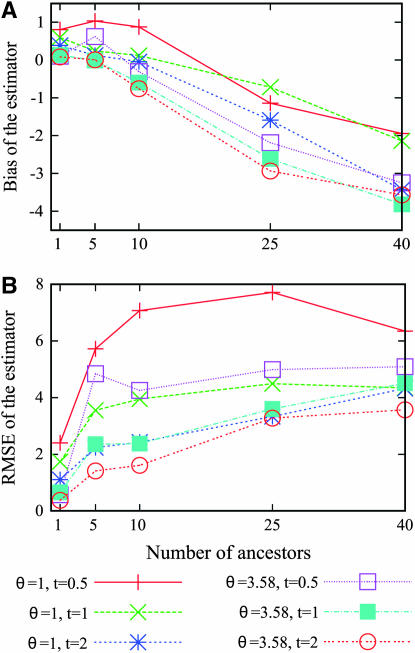
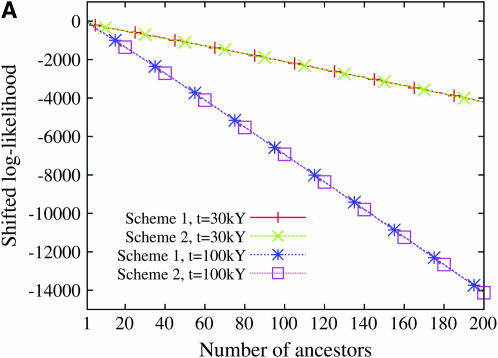
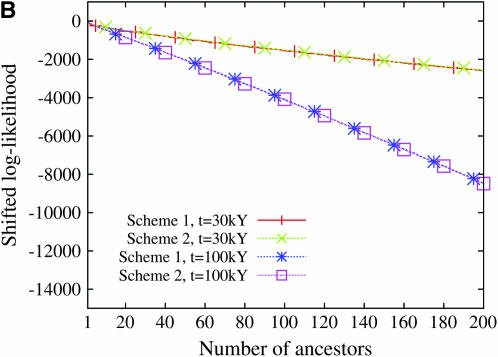
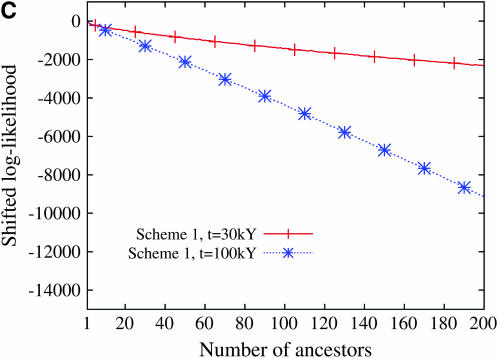
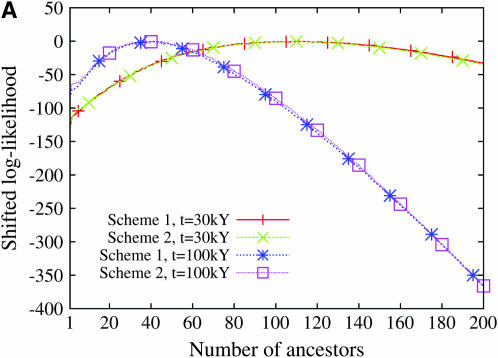
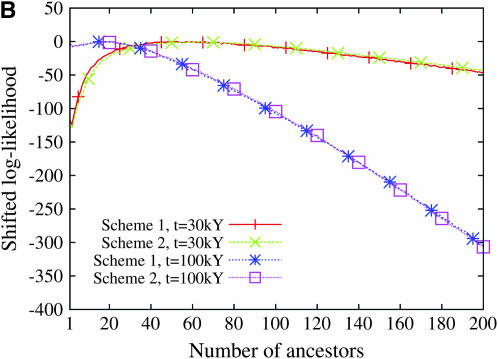
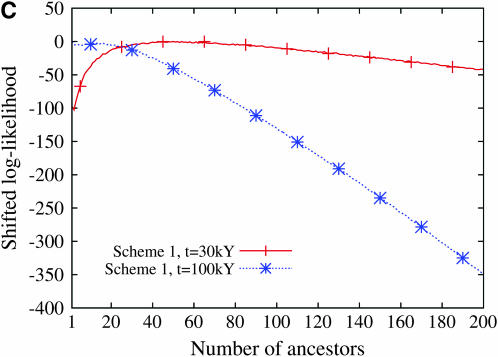
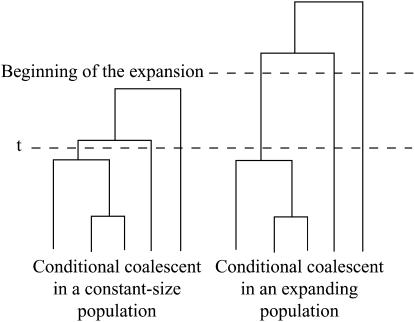
Estimating the number of ancestral lineages of a sample of DNA sequences at time t in the past can be viewed as a variation on the problem of estimating the time to the most recent common ancestor. To estimate the number of ancestral lineages, we develop a maximum-likelihood approach that takes advantage of a prior model of population demography, in addition to the molecular data summarized by the pattern of polymorphic sites. The method relies on a rejection sampling algorithm that is introduced for simulating conditional coalescent trees given a fixed number of ancestral lineages at time t. Computer simulations show that the number of ancestral lineages can be estimated accurately, provided that the number of mutations that occurred since time t is sufficiently large. The method is applied to 986 present-day human sequences located in hypervariable region 1 of the mitochondrion to estimate the number of ancestral lineages of modern humans at the time of potential admixture with the Neanderthal population. Our estimates support a view that the proportion of the modern population consisting of Neanderthal contributions must be relatively small, less than approximately 5%, if the admixture happened as recently as 30,000 years ago.
Figures
References
-
- Bignami, A., and A. De Matteis, 1971. A note on sampling from combinations of distributions. IMA J. Appl. Math. 8: 80–81.
-
- Biraben, J.-N., 1979. Essai sur l'évolution du nombre des hommes. Population 1: 13–25.
-
- Blum, M. G. B., and O. François, 2005. On statistical tests of phylogenetic tree imbalance: the Sackin and other indices revisited. Math. Biosci. 195: 141–153. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
