

A Bayesian multilocus association method: allowing for higher-order interaction in association studies
- PMID: 17435250
- PMCID: PMC1894584
- DOI: 10.1534/genetics.107.071696
A Bayesian multilocus association method: allowing for higher-order interaction in association studies
Abstract
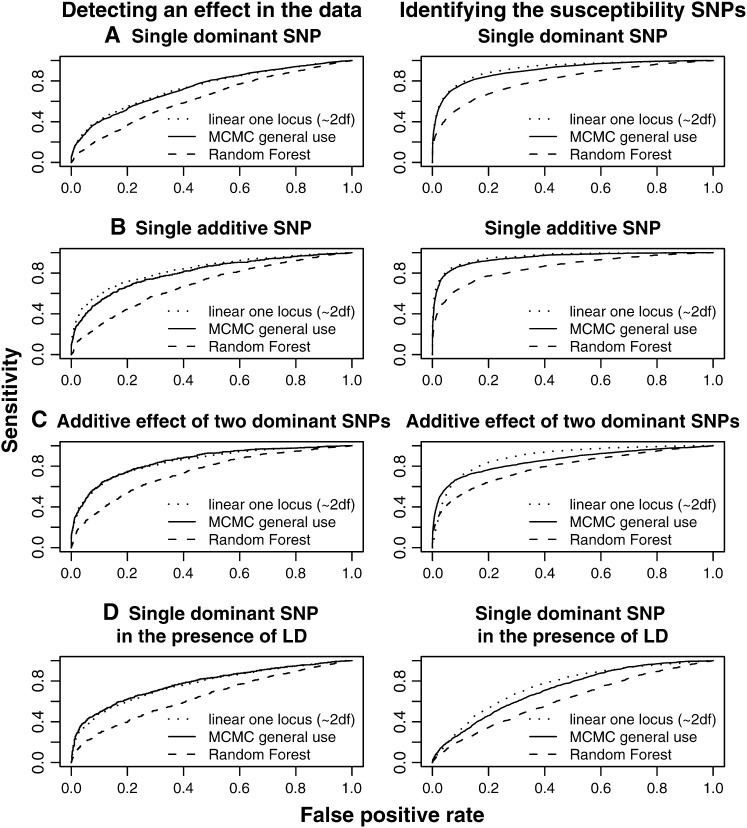
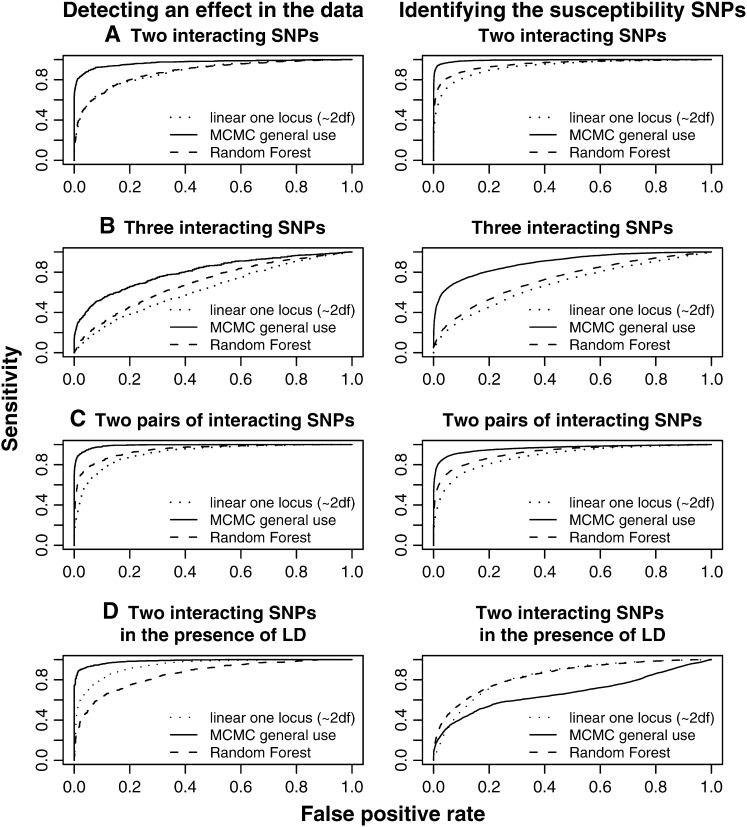
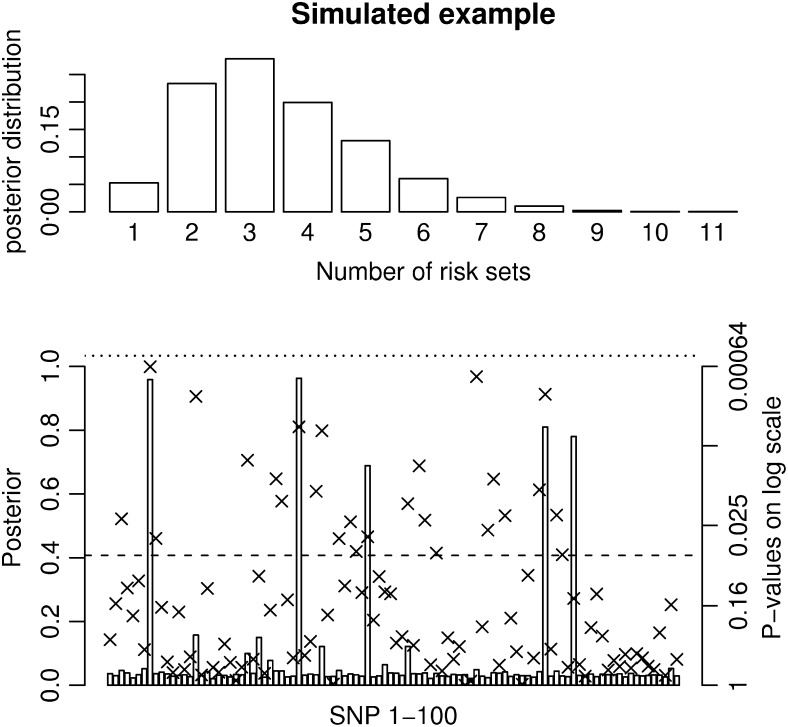
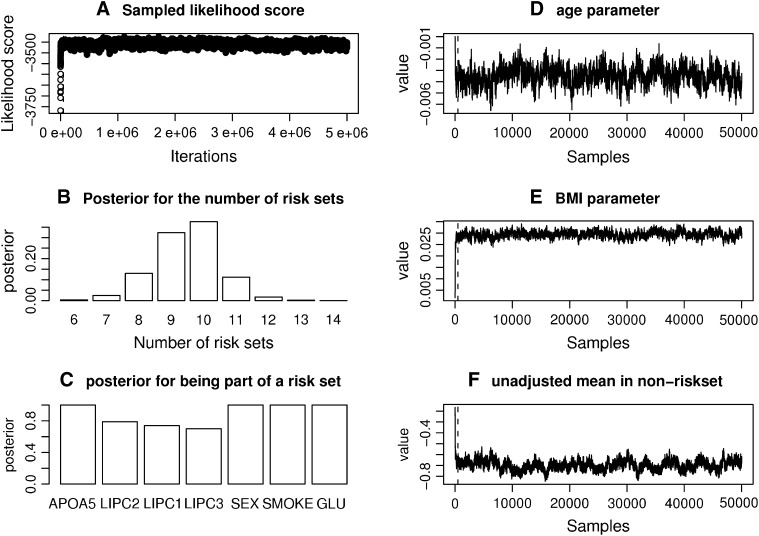
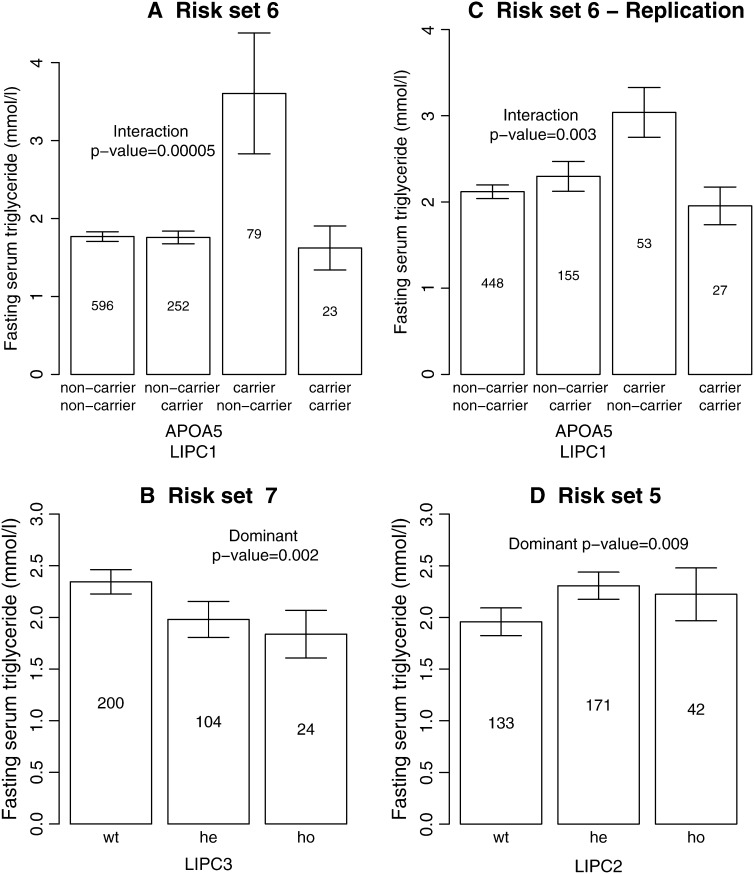
For most common diseases with heritable components, not a single or a few single-nucleotide polymorphisms (SNPs) explain most of the variance for these disorders. Instead, much of the variance may be caused by interactions (epistasis) among multiple SNPs or interactions with environmental conditions. We present a new powerful statistical model for analyzing and interpreting genomic data that influence multifactorial phenotypic traits with a complex and likely polygenic inheritance. The new method is based on Markov chain Monte Carlo (MCMC) and allows for identification of sets of SNPs and environmental factors that when combined increase disease risk or change the distribution of a quantitative trait. Using simulations, we show that the MCMC method can detect disease association when multiple, interacting SNPs are present in the data. When applying the method on real large-scale data from a Danish population-based cohort, multiple interactions are identified that severely affect serum triglyceride levels in the study individuals. The method is designed for quantitative traits but can also be applied on qualitative traits. It is computationally feasible even for a large number of possible interactions and differs fundamentally from most previous approaches by entertaining nonlinear interactions and by directly addressing the multiple-testing problem.
Figures
References
-
- Baker, S. G., 2005. A simple loglinear model for haplotype effects in a case-control study involving two unphased genotypes. Stat. Appl. Genet. Mol. Biol. 4 14. - PubMed
-
- Bellman, R. E., 1961. Adaptive Control Processes. Princeton University Press, Princeton, NJ.
-
- Breiman, L., 2001. Random forest. Mach. Learn. 45 5–32.
-
- Breiman, L., J. H. Friedman, R. A. Olshen and C. J. Stone, 1984. Classification and Regression Trees, Ed. 1. Wadsworth, Belmont, CA.
-
- Brooks, S. P., and A. Gelman, 1998. General methods for monitoring convergence of iteractive simulations. J. Comp. Graph. Stat. 7 434–455.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
