

Mutations in cytochrome c oxidase subunit VIa cause neurodegeneration and motor dysfunction in Drosophila
- PMID: 17435251
- PMCID: PMC1894620
- DOI: 10.1534/genetics.107.071688
Mutations in cytochrome c oxidase subunit VIa cause neurodegeneration and motor dysfunction in Drosophila
Abstract
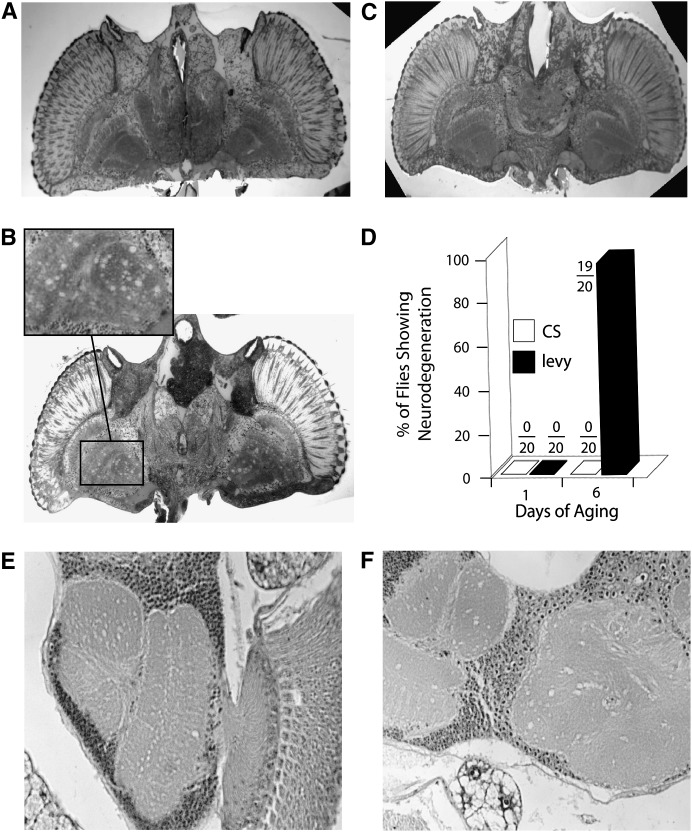
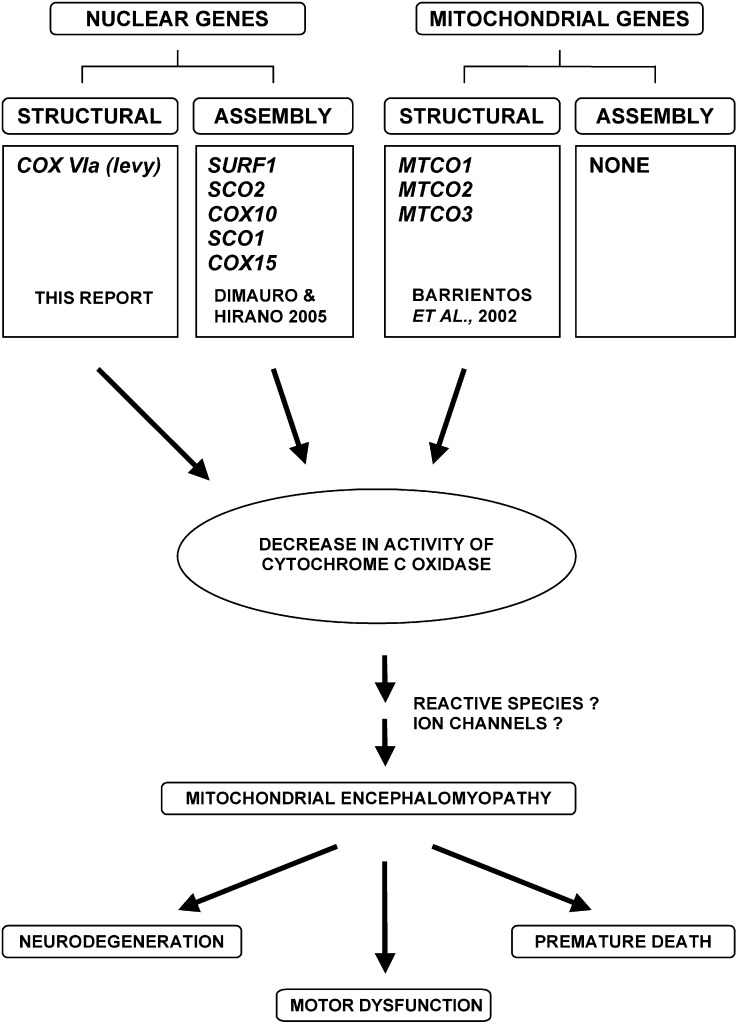
Mitochondrial dysfunction is involved in many neurodegenerative disorders in humans. Here we report mutations in a gene (designated levy) that codes for subunit VIa of cytochrome c oxidase (COX). The mutations were identified by the phenotype of temperature-induced paralysis and showed the additional phenotypes of decreased COX activity, age-dependent bang-induced paralysis, progressive neurodegeneration, and reduced life span. Germ-line transformation using the levy(+) gene rescued the mutant flies from all phenotypes including neurodegeneration. The data from levy mutants reveal a COX-mediated pathway in Drosophila, disruption of which leads to mitochondrial encephalomyopathic effects including neurodegeneration, motor dysfunction, and premature death. The data present the first case of a mutation in a nuclear-encoded structural subunit of COX that causes mitochondrial encephalomyopathy rather than lethality, whereas several previous attempts to identify such mutations have not been successful. The levy mutants provide a genetic model to understand the mechanisms underlying COX-mediated mitochondrial encephalomyopathies and to explore possible therapeutic interventions.
Figures
Similar articles
-
Genetic mosaic analysis of a deleterious mitochondrial DNA mutation in Drosophila reveals novel aspects of mitochondrial regulation and function.Mol Biol Cell. 2015 Feb 15;26(4):674-84. doi: 10.1091/mbc.E14-11-1513. Epub 2014 Dec 10. Mol Biol Cell. 2015. PMID: 25501370 Free PMC article.
-
Polyglutamine tract-binding protein-1 dysfunction induces cell death of neurons through mitochondrial stress.J Neurochem. 2005 Nov;95(3):858-70. doi: 10.1111/j.1471-4159.2005.03405.x. Epub 2005 Aug 16. J Neurochem. 2005. PMID: 16104847
-
Temperature-sensitive paralytic mutants are enriched for those causing neurodegeneration in Drosophila.Genetics. 2002 Jul;161(3):1197-208. doi: 10.1093/genetics/161.3.1197. Genetics. 2002. PMID: 12136022 Free PMC article.
-
Human mitochondrial COX1 assembly into cytochrome c oxidase at a glance.J Cell Sci. 2015 Mar 1;128(5):833-7. doi: 10.1242/jcs.161729. Epub 2015 Feb 6. J Cell Sci. 2015. PMID: 25663696 Review.
-
The subunit composition and function of mammalian cytochrome c oxidase.Mitochondrion. 2015 Sep;24:64-76. doi: 10.1016/j.mito.2015.07.002. Epub 2015 Jul 17. Mitochondrion. 2015. PMID: 26190566 Review.
Cited by
-
LACK, a RACK1 ortholog, facilitates cytochrome c oxidase subunit expression to promote Leishmania major fitness.Mol Microbiol. 2015 Apr;96(1):95-109. doi: 10.1111/mmi.12924. Epub 2015 Feb 4. Mol Microbiol. 2015. PMID: 25582232 Free PMC article.
-
Mitochondrial ROS and the Effectors of the Intrinsic Apoptotic Pathway in Aging Cells: The Discerning Killers!Front Genet. 2016 Sep 14;7:161. doi: 10.3389/fgene.2016.00161. eCollection 2016. Front Genet. 2016. PMID: 27683586 Free PMC article. Review.
-
Maintaining the brain: insight into human neurodegeneration from Drosophila melanogaster mutants.Nat Rev Genet. 2009 Jun;10(6):359-70. doi: 10.1038/nrg2563. Nat Rev Genet. 2009. PMID: 19434080 Free PMC article. Review.
-
SDHAF4 promotes mitochondrial succinate dehydrogenase activity and prevents neurodegeneration.Cell Metab. 2014 Aug 5;20(2):241-52. doi: 10.1016/j.cmet.2014.05.012. Epub 2014 Jun 19. Cell Metab. 2014. PMID: 24954416 Free PMC article.
-
Severe infantile encephalomyopathy caused by a mutation in COX6B1, a nucleus-encoded subunit of cytochrome c oxidase.Am J Hum Genet. 2008 Jun;82(6):1281-9. doi: 10.1016/j.ajhg.2008.05.002. Epub 2008 May 22. Am J Hum Genet. 2008. PMID: 18499082 Free PMC article.
References
-
- Abou-Sleiman, P. M., M. M. Muqit and N. W. Wood, 2006. Expanding insights of mitochondrial dysfunction in Parkinson's disease. Nat. Rev. Neurosci. 7 207–219. - PubMed
-
- Agostino, A., F. Invernizzi, C. Tiveron, G. Fagiolari, A. Prelle et al., 2003. Constitutive knockout of Surf1 is associated with high embryonic lethality, mitochondrial disease and cytochrome c oxidase deficiency in mice. Hum. Mol. Genet. 12 399–413. - PubMed
-
- Ashburner, M., 1989. Drosophila: A Laboratory Handbook. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
-
- Barrientos, A., M. H. Barros, I. Valnot, A. Rotig, P. Rustin et al., 2002. Cytochrome oxidase in health and disease. Gene 286 53–63. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
