

Astrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons
- PMID: 17435755
- PMCID: PMC3799799
- DOI: 10.1038/nn1876
Astrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons
Abstract
Mutations in superoxide dismutase-1 (SOD1) cause a form of the fatal paralytic disorder amyotrophic lateral sclerosis (ALS), presumably by a combination of cell-autonomous and non-cell-autonomous processes. Here, we show that expression of mutated human SOD1 in primary mouse spinal motor neurons does not provoke motor neuron degeneration. Conversely, rodent astrocytes expressing mutated SOD1 kill spinal primary and embryonic mouse stem cell-derived motor neurons. This is triggered by soluble toxic factor(s) through a Bax-dependent mechanism. However, mutant astrocytes do not cause the death of spinal GABAergic or dorsal root ganglion neurons or of embryonic stem cell-derived interneurons. In contrast to astrocytes, fibroblasts, microglia, cortical neurons and myocytes expressing mutated SOD1 do not cause overt neurotoxicity. These findings indicate that astrocytes may play a role in the specific degeneration of spinal motor neurons in ALS. Identification of the astrocyte-derived soluble factor(s) may have far-reaching implications for ALS from both a pathogenic and therapeutic standpoint.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
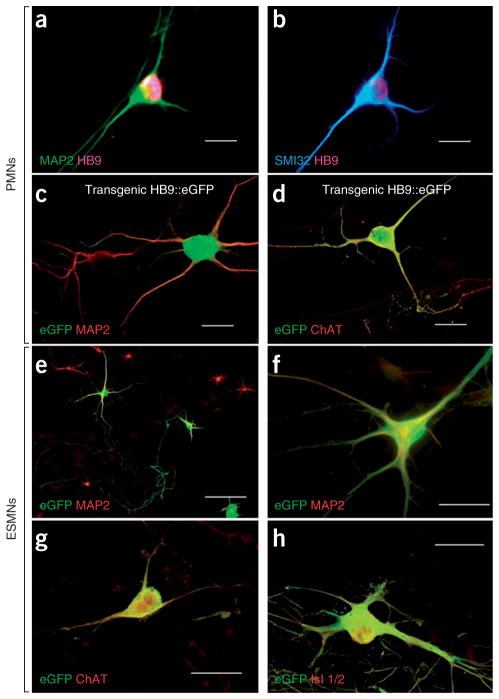
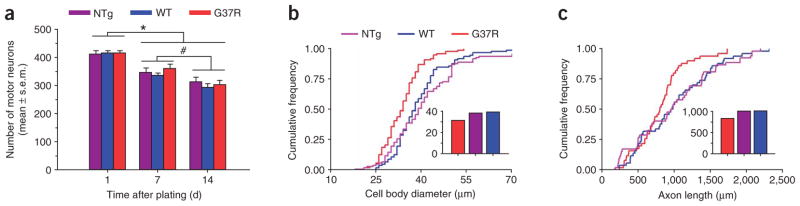
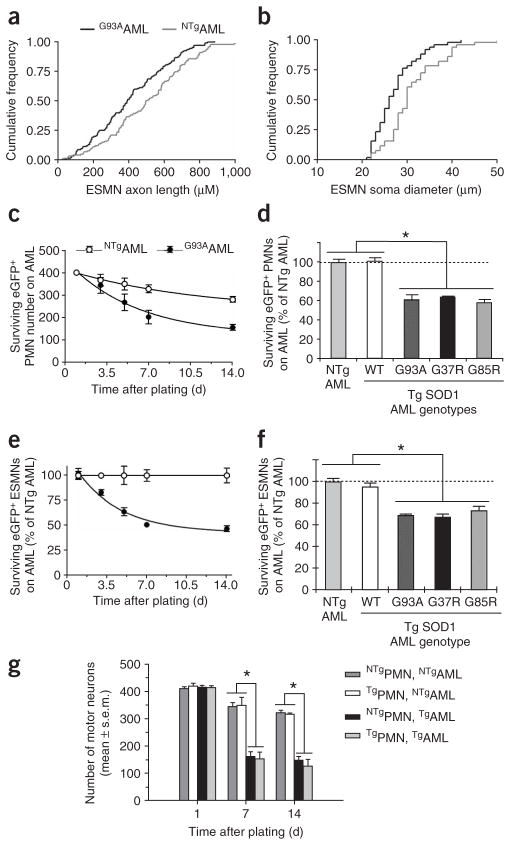
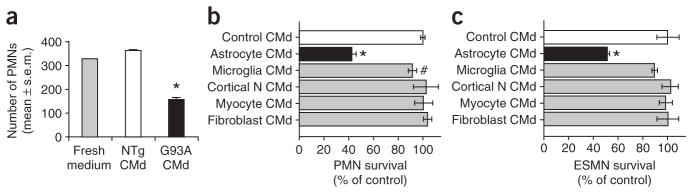
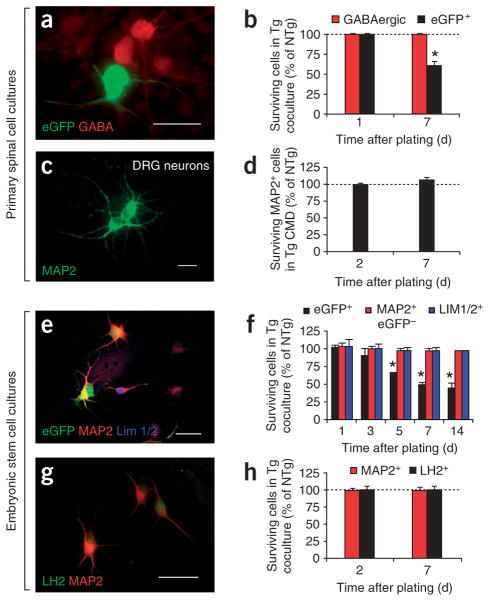
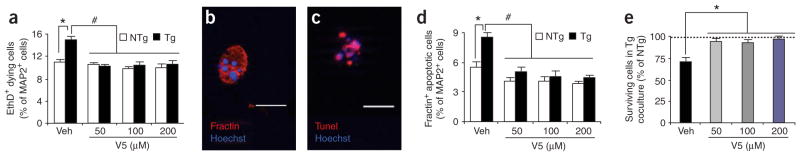
Comment in
-
ALS: astrocytes move in as deadly neighbors.Nat Neurosci. 2007 May;10(5):535-7. doi: 10.1038/nn0507-535. Nat Neurosci. 2007. PMID: 17453052 No abstract available.
-
Astrocytes usurp neurons as a disease focus.Nat Neurosci. 2019 Apr;22(4):512-513. doi: 10.1038/s41593-019-0367-6. Nat Neurosci. 2019. PMID: 30858602 No abstract available.
References
-
- Rosen DR, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62. - PubMed
-
- Deng HX, et al. Amyotrophic lateral sclerosis and structural defects in Cu,Zn superoxide dismutase. Science. 1993;261:1047–1051. - PubMed
-
- Gurney ME, et al. Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase mutation. Science. 1994;264:1772–1775. - PubMed
-
- Wong PC, et al. An adverse property of a familial ALS-linked SOD1 mutation causes motor neuron disease characterized by vacuolar degeneration of mitochondria. Neuron. 1995;14:1105–1116. - PubMed
-
- Bruijn LI, et al. ALS-linked SOD1 mutant G85R mediated damage to astrocytes and promotes rapidly progressive disease with SOD1-containing inclusions. Neuron. 1997;18:327–338. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
