

Hypomorphic mutations in the gene encoding a key Fanconi anemia protein, FANCD2, sustain a significant group of FA-D2 patients with severe phenotype
- PMID: 17436244
- PMCID: PMC1852747
- DOI: 10.1086/517616
Hypomorphic mutations in the gene encoding a key Fanconi anemia protein, FANCD2, sustain a significant group of FA-D2 patients with severe phenotype
Erratum in
- Am J Hum Genet. 2007 Jul;81(1):196
Abstract
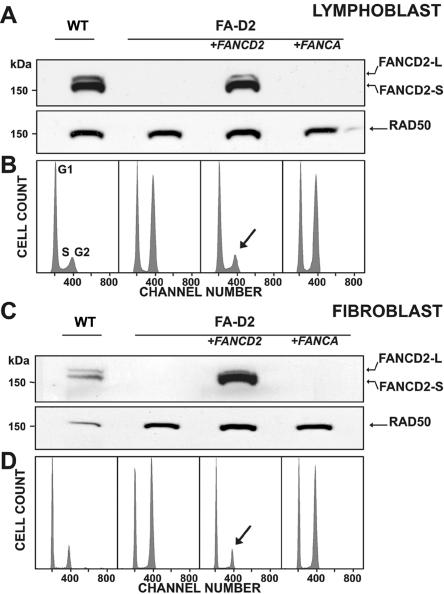
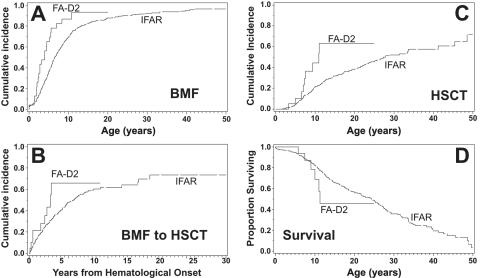
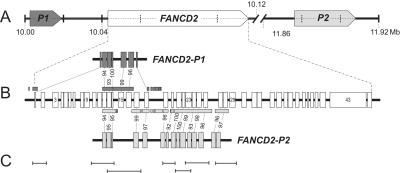
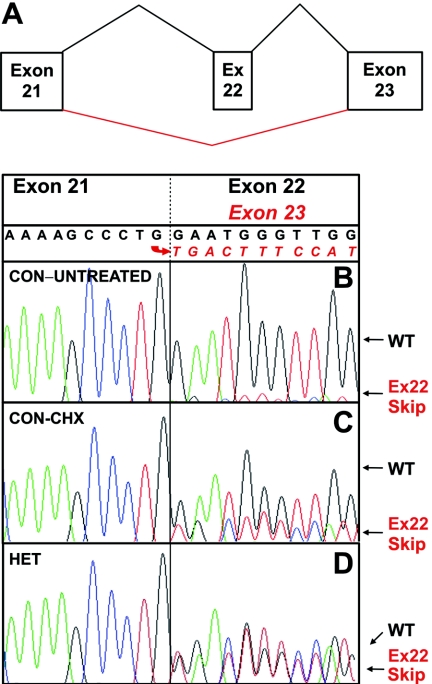
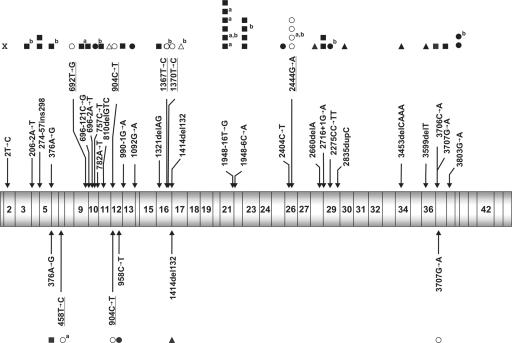
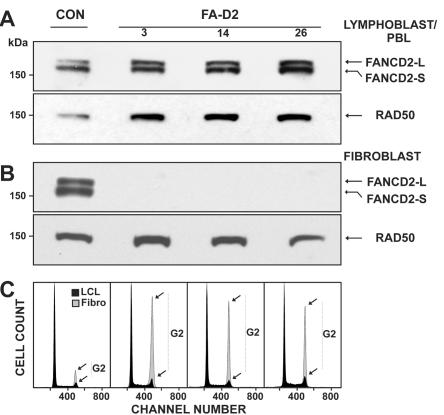
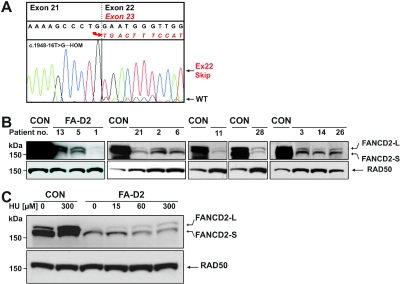
FANCD2 is an evolutionarily conserved Fanconi anemia (FA) gene that plays a key role in DNA double-strand-type damage responses. Using complementation assays and immunoblotting, a consortium of American and European groups assigned 29 patients with FA from 23 families and 4 additional unrelated patients to complementation group FA-D2. This amounts to 3%-6% of FA-affected patients registered in various data sets. Malformations are frequent in FA-D2 patients, and hematological manifestations appear earlier and progress more rapidly when compared with all other patients combined (FA-non-D2) in the International Fanconi Anemia Registry. FANCD2 is flanked by two pseudogenes. Mutation analysis revealed the expected total of 66 mutated alleles, 34 of which result in aberrant splicing patterns. Many mutations are recurrent and have ethnic associations and shared allelic haplotypes. There were no biallelic null mutations; residual FANCD2 protein of both isotypes was observed in all available patient cell lines. These analyses suggest that, unlike the knockout mouse model, total absence of FANCD2 does not exist in FA-D2 patients, because of constraints on viable combinations of FANCD2 mutations. Although hypomorphic mutations arie involved, clinically, these patients have a relatively severe form of FA.
Figures
References
Web Resources
-
- ClustalW, ftp://ftp-igbmc.u-strasbg.fr/pub/ClustalX (for the polypeptide sequences compared using the Windows interface ClustalX [v. 1.81, for the multiple sequence-alignment program])
-
- CpG Island Explorer, http://bioinfo.hku.hk/cpgieintro.html (for promoter analyses [v. 1.9, at the settings GC 60%, CpG O/E ratio 0.7, and minimum length 500 nt)
-
- Ensembl Genome Browser, http://www.ensembl.org/ (for FANCD2 genomic sequences [accession number ENSG00000144554] and Fancd2 sequence information for other species)
-
- ESEfinder, http://rulai.cshl.edu/tools/ESE/ (for analysis of regulatory splice sequences)
-
- ExPASy, http://www.expasy.org/sprot/ (for the Swiss-Prot Fancd2 protein sequences of different species, including Homo sapiens)
References
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
