

Growth control of the eukaryote cell: a systems biology study in yeast
- PMID: 17439666
- PMCID: PMC2373899
- DOI: 10.1186/jbiol54
Growth control of the eukaryote cell: a systems biology study in yeast
Abstract
Background: Cell growth underlies many key cellular and developmental processes, yet a limited number of studies have been carried out on cell-growth regulation. Comprehensive studies at the transcriptional, proteomic and metabolic levels under defined controlled conditions are currently lacking.
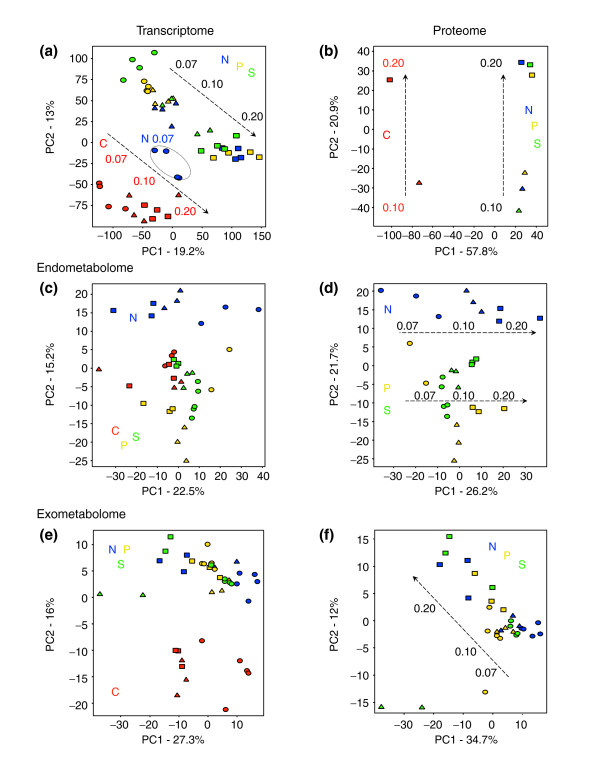
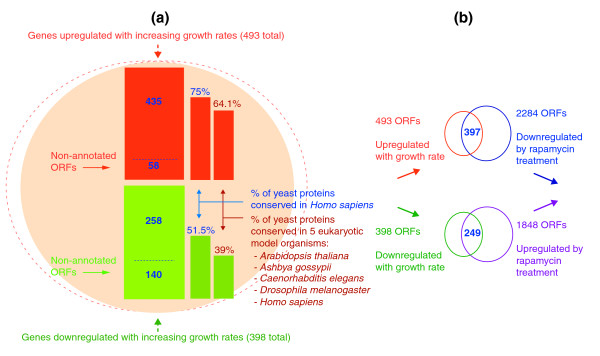
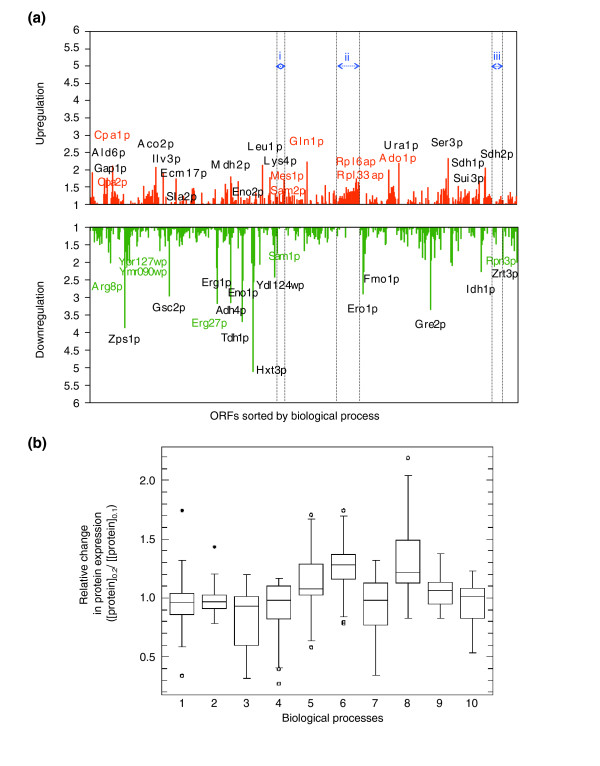
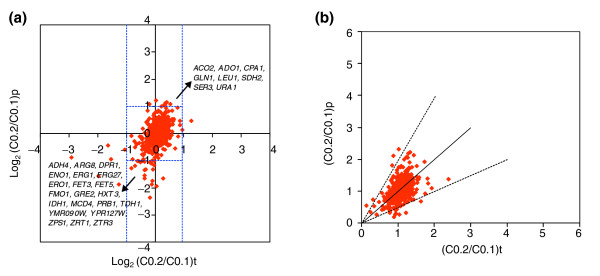
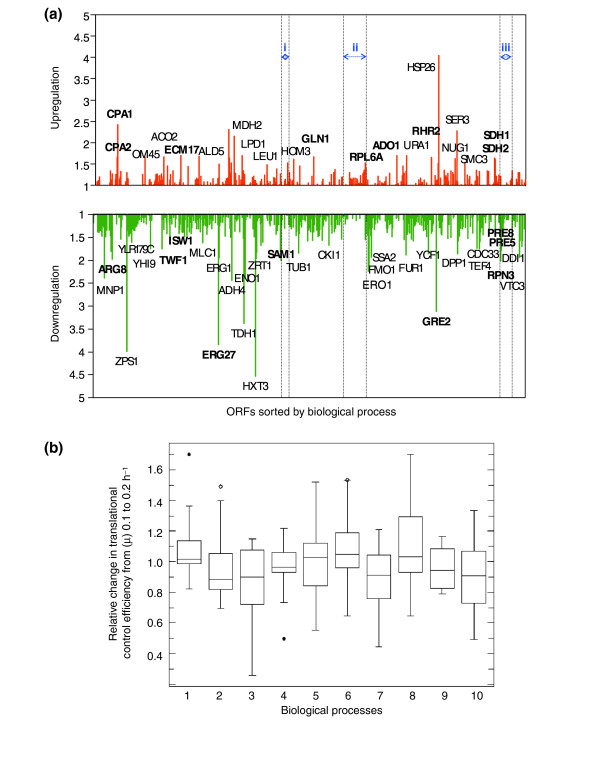
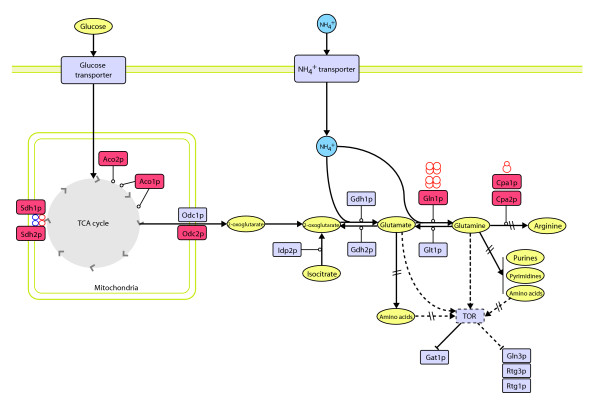
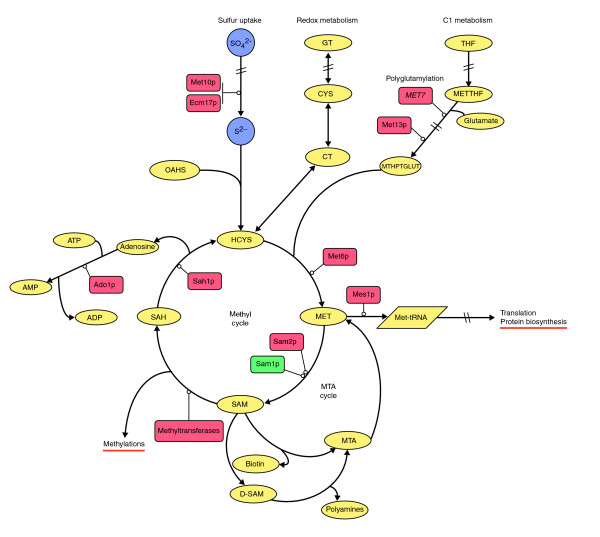
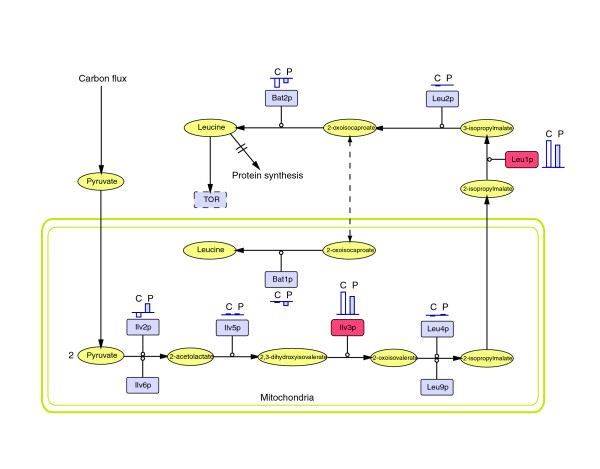
Results: Metabolic control analysis is being exploited in a systems biology study of the eukaryotic cell. Using chemostat culture, we have measured the impact of changes in flux (growth rate) on the transcriptome, proteome, endometabolome and exometabolome of the yeast Saccharomyces cerevisiae. Each functional genomic level shows clear growth-rate-associated trends and discriminates between carbon-sufficient and carbon-limited conditions. Genes consistently and significantly upregulated with increasing growth rate are frequently essential and encode evolutionarily conserved proteins of known function that participate in many protein-protein interactions. In contrast, more unknown, and fewer essential, genes are downregulated with increasing growth rate; their protein products rarely interact with one another. A large proportion of yeast genes under positive growth-rate control share orthologs with other eukaryotes, including humans. Significantly, transcription of genes encoding components of the TOR complex (a major controller of eukaryotic cell growth) is not subject to growth-rate regulation. Moreover, integrative studies reveal the extent and importance of post-transcriptional control, patterns of control of metabolic fluxes at the level of enzyme synthesis, and the relevance of specific enzymatic reactions in the control of metabolic fluxes during cell growth.
Conclusion: This work constitutes a first comprehensive systems biology study on growth-rate control in the eukaryotic cell. The results have direct implications for advanced studies on cell growth, in vivo regulation of metabolic fluxes for comprehensive metabolic engineering, and for the design of genome-scale systems biology models of the eukaryotic cell.
Figures
References
-
- Kacser H, Burns JA. The control of flux. Symp Soc Exp Biol. 1973;27:65–104. - PubMed
-
- Warner JR. The economics of ribosome biosynthesis in yeast. Trends Biochem Sci. 1999;24:437–440. - PubMed
-
- Fingar DC, Blenis J. Target of rapamycin (TOR): an integrator of nutrient and growth factor signals and coordinator of cell growth and cell cycle progression. Oncogene. 2004;23:3151–3171. - PubMed
-
- Hall MN, Raff M, Thomas G, Eds . Cell Growth: Control of Cell Size Monograph 42. New York: Cold Spring Harbor Laboratory Press; 2004.
-
- Jorgensen P, Tyers M, Warner JR. Forging the factory: ribosome synthesis and growth control in budding yeast. In: Hall MN, Raff M, Thomas G, editor. Cell Growth: Control of Cell Size Monograph 42. New York: Cold Spring Harbor Laboratory Press; 2004. pp. 329–370.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
