

A new algorithm using cross-assignment for label-free quantitation with LC-LTQ-FT MS
- PMID: 17441747
- PMCID: PMC2563808
- DOI: 10.1021/pr0606880
A new algorithm using cross-assignment for label-free quantitation with LC-LTQ-FT MS
Abstract
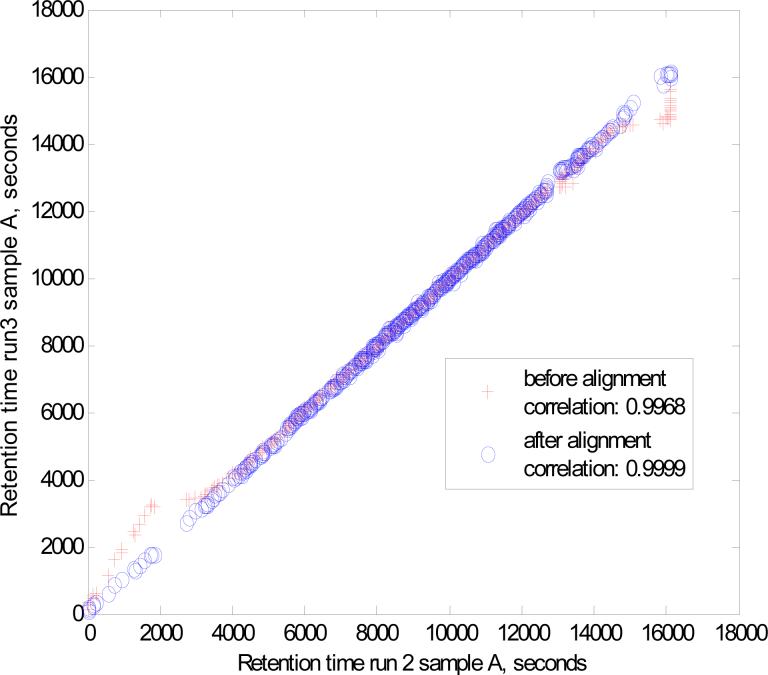
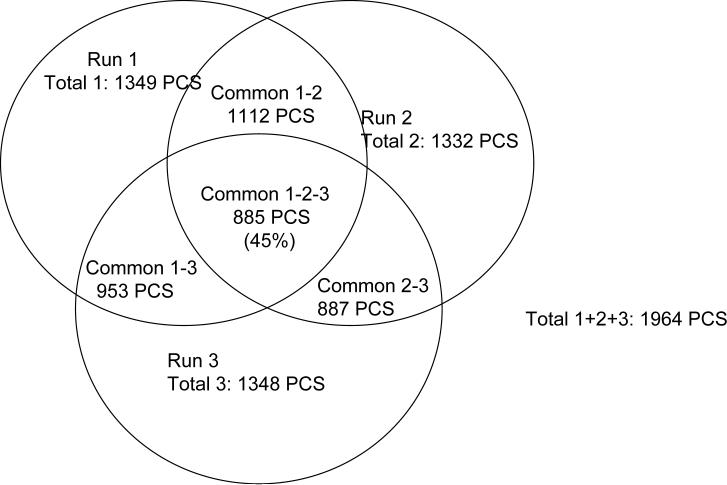
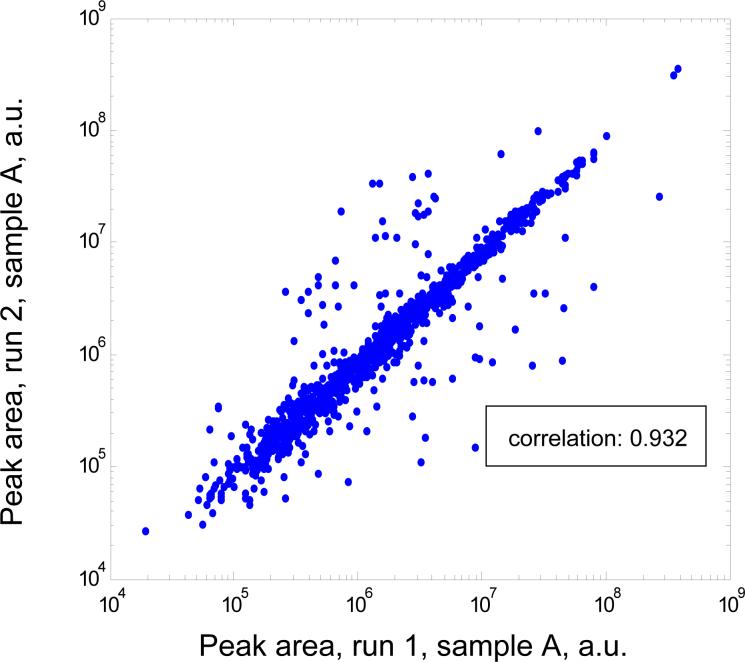
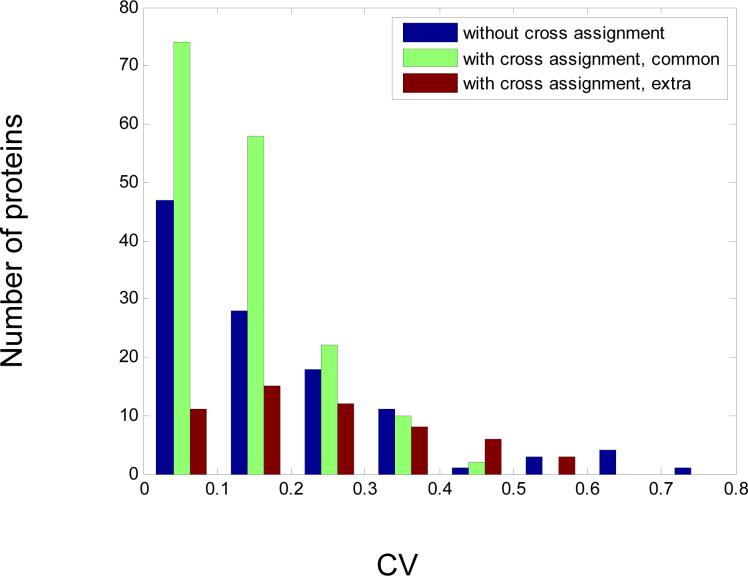
A new algorithm is described for label-free quantitation of relative protein abundances across multiple complex proteomic samples. Q-MEND is based on the denoising and peak picking algorithm, MEND, previously developed in our laboratory. Q-MEND takes advantage of the high resolution and mass accuracy of the hybrid LTQ-FT MS mass spectrometer (or other high-resolution mass spectrometers, such as a Q-TOF MS). The strategy, termed "cross-assignment", is introduced to increase substantially the number of quantitated proteins. In this approach, all MS/MS identifications for the set of analyzed samples are combined into a master ID list, and then each LC-MS run is searched for the features that can be assigned to a specific identification from that master list. The reliability of quantitation is enhanced by quantitating separately all peptide charge states, along with a scoring procedure to filter out less reliable peptide abundance measurements. The effectiveness of Q-MEND is illustrated in the relative quantitative analysis of Escherichia coli samples spiked with known amounts of non-E. coli protein digests. A mean quantitation accuracy of 7% and mean precision of 15% is demonstrated. Q-MEND can perform relative quantitation of a set of LC-MS data sets without manual intervention and can generate files compatible with the Guidelines for Proteomic Data Publication.
Figures


References
-
- Ong S-E, Mann M. Mass spectrometry-based proteomics turns quantitative. Nat. Chem. Biol. 2005;1:252–262. - PubMed
-
- Gygi SP, Rist B, Gerber SA, Turecek F, Gelb MH, Aebersold R. Quantitative analysis of complex protein mixtures using isotope- coded affinity tags. Nat. Biotechnol. 1999;17:994–999. - PubMed
-
- Washburn MP, Ulaszek R, Deciu C, Schieltz DM, Yates JR. Analysis of quantitative proteomic data generated via multidimensional protein identification technology. Anal. Chem. 2002;74:1650–1657. - PubMed
-
- Schulze WX, Mann M. A novel proteomic screen for peptide-protein interactions. J. Biol. Chem. 2003;279:10756–10764. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
