

Efficient pairwise RNA structure prediction using probabilistic alignment constraints in Dynalign
- PMID: 17445273
- PMCID: PMC1868766
- DOI: 10.1186/1471-2105-8-130
Efficient pairwise RNA structure prediction using probabilistic alignment constraints in Dynalign
Abstract
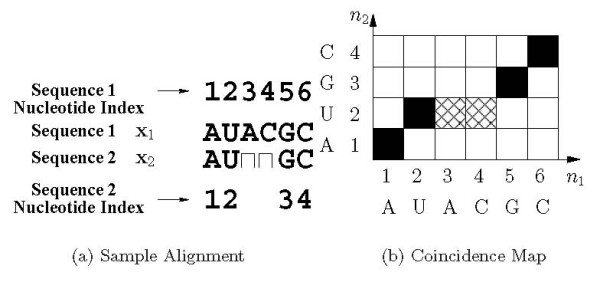
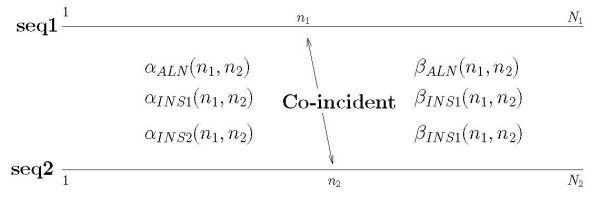
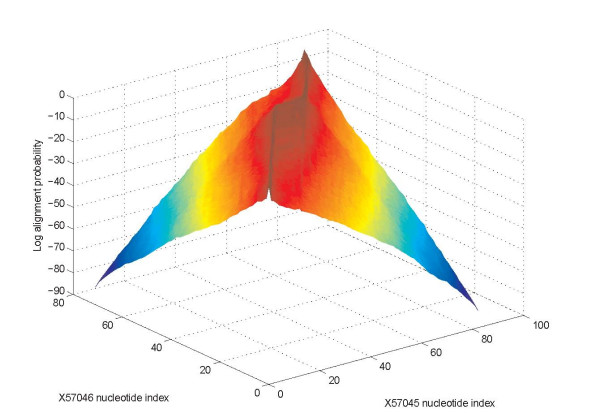
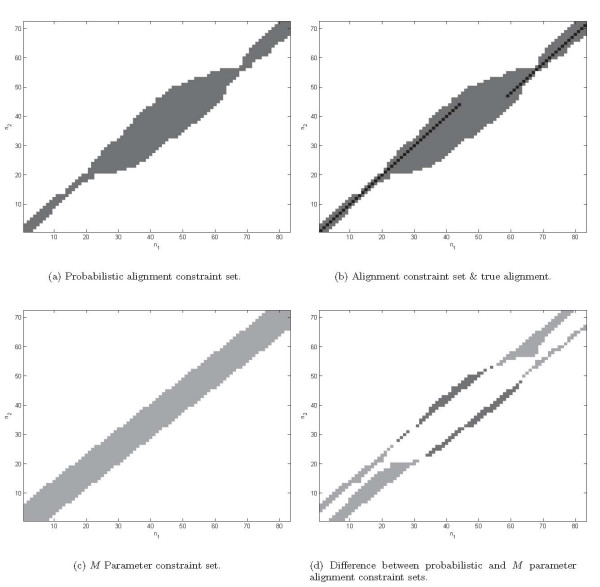
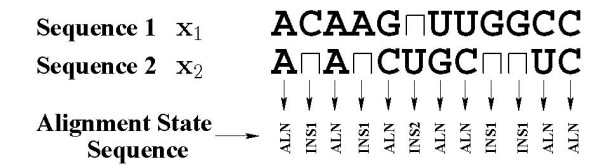
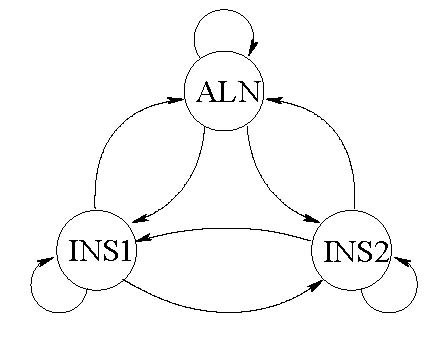
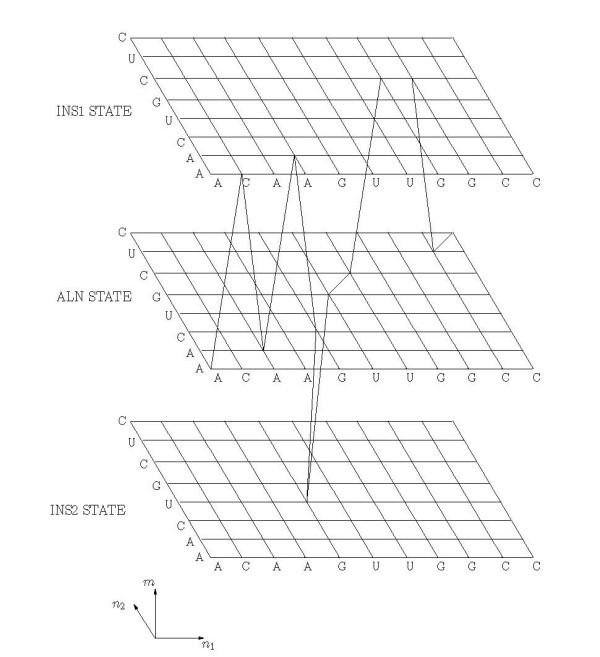
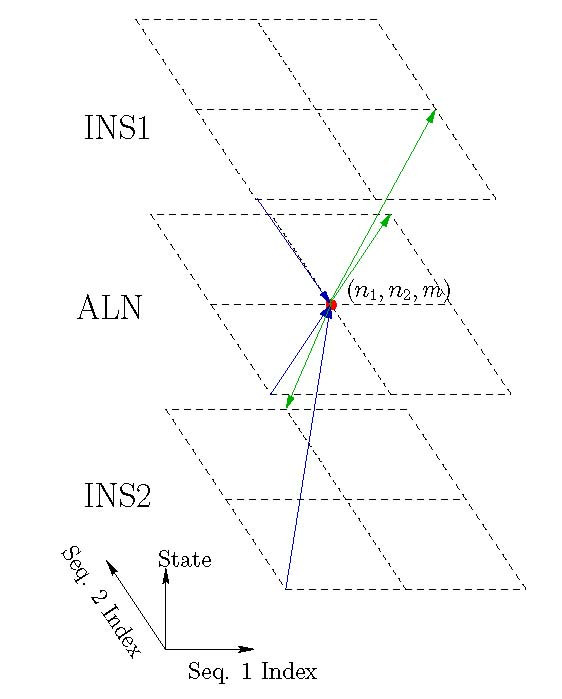
Background: Joint alignment and secondary structure prediction of two RNA sequences can significantly improve the accuracy of the structural predictions. Methods addressing this problem, however, are forced to employ constraints that reduce computation by restricting the alignments and/or structures (i.e. folds) that are permissible. In this paper, a new methodology is presented for the purpose of establishing alignment constraints based on nucleotide alignment and insertion posterior probabilities. Using a hidden Markov model, posterior probabilities of alignment and insertion are computed for all possible pairings of nucleotide positions from the two sequences. These alignment and insertion posterior probabilities are additively combined to obtain probabilities of co-incidence for nucleotide position pairs. A suitable alignment constraint is obtained by thresholding the co-incidence probabilities. The constraint is integrated with Dynalign, a free energy minimization algorithm for joint alignment and secondary structure prediction. The resulting method is benchmarked against the previous version of Dynalign and against other programs for pairwise RNA structure prediction.
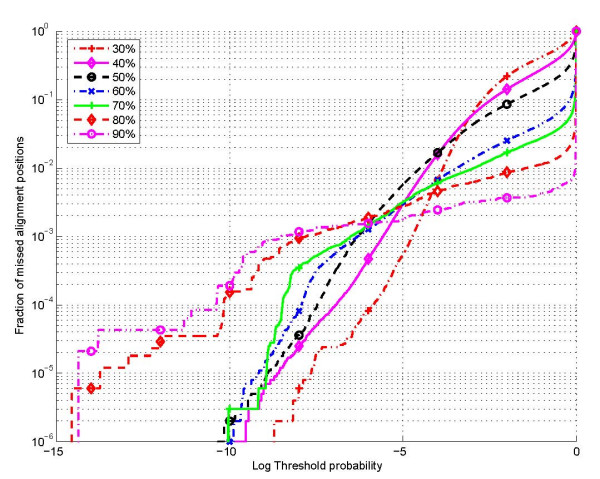
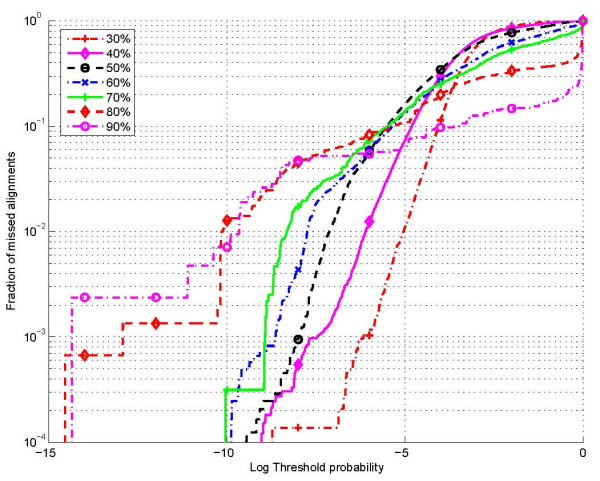
Results: The proposed technique eliminates manual parameter selection in Dynalign and provides significant computational time savings in comparison to prior constraints in Dynalign while simultaneously providing a small improvement in the structural prediction accuracy. Savings are also realized in memory. In experiments over a 5S RNA dataset with average sequence length of approximately 120 nucleotides, the method reduces computation by a factor of 2. The method performs favorably in comparison to other programs for pairwise RNA structure prediction: yielding better accuracy, on average, and requiring significantly lesser computational resources.
Conclusion: Probabilistic analysis can be utilized in order to automate the determination of alignment constraints for pairwise RNA structure prediction methods in a principled fashion. These constraints can reduce the computational and memory requirements of these methods while maintaining or improving their accuracy of structural prediction. This extends the practical reach of these methods to longer length sequences. The revised Dynalign code is freely available for download.
Figures
References
-
- Durbin R, Eddy SR, Krogh A, Mitchison G. Biological Sequence Analysis : Probabilistic Models of Proteins and Nucleic Acids. Cambridge, UK: Cambridge University Press; 1999.
-
- Pace NR, Thomas BC, Woese CR. The RNA World. second. Cold Spring Harbor Laboratory Press; 1999. Probing RNA structure, function and history by comparative analysis; pp. 113–141.
-
- Sankoff D. Simultaneous Solution of RNA Folding, Alignment and Protosequence Problems. SIAM J App Math. 1985;8(5):810–825. doi: 10.1137/0145048. - DOI
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
