

Activation of MAPK pathways links LMNA mutations to cardiomyopathy in Emery-Dreifuss muscular dystrophy
- PMID: 17446932
- PMCID: PMC1849984
- DOI: 10.1172/JCI29042
Activation of MAPK pathways links LMNA mutations to cardiomyopathy in Emery-Dreifuss muscular dystrophy
Abstract
Mutations in LMNA, which encodes nuclear Lamins A and C cause diseases affecting various organs, including the heart. We have determined the effects of an Lmna H222P mutation on signaling pathways involved in the development of cardiomyopathy in a knockin mouse model of autosomal dominant Emery-Dreifuss muscular dystrophy. Analysis of genome-wide expression profiles in hearts using Affymetrix GeneChips showed statistically significant differences in expression of genes in the MAPK pathways at the incipience of the development of clinical disease. Using real-time PCR, we showed that activation of MAPK pathways preceded clinical signs or detectable molecular markers of cardiomyopathy. In heart tissue and isolated cardiomyocytes, there was activation of MAPK cascades and downstream targets, implicated previously in the pathogenesis of cardiomyopathy. Expression of H222P Lamin A in cultured cells activated MAPKs and downstream target genes. Activation of MAPK signaling by mutant A-type lamins could be a cornerstone in the development of heart disease in autosomal dominant Emery-Dreifuss muscular dystrophy.
Figures



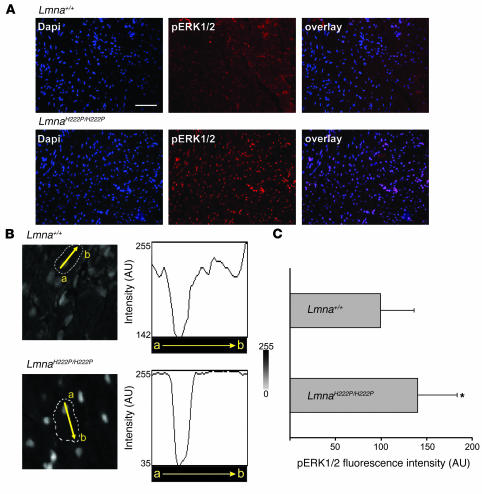





References
-
- Emery A.E.H. Emery-Dreifuss muscular dystrophy — a 40 year retrospective. Neuromusc. Disord. . 2000;10:228–232. - PubMed
-
- Bione S., et al. Identification of a novel X-linked gene responsible for Emery-Dreifuss muscular dystrophy. Nat. Genet. . 1994;8:323–327. - PubMed
-
- Manilal S., Nguyen T.M., Sewry C.A., Morris G.E. The Emery-Dreifuss muscular dystrophy protein, emerin, is a nuclear membrane protein. Hum. Mol. Genet. . 1996;5:801–808. - PubMed
-
- Nagano A., et al. Emerin deficiency at the nuclear membrane in patients with Emery-Dreifuss muscular dystrophy. Nat. Genet. . 1996;12:254–259. - PubMed
-
- Bonne G., et al. Mutations in the gene encoding lamin A/C cause autosomal dominant Emery-Dreifuss muscular dystrophy. Nat. Genet. . 1999;21:285–288. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
