

Supervised multivariate analysis of sequence groups to identify specificity determining residues
- PMID: 17451607
- PMCID: PMC1878507
- DOI: 10.1186/1471-2105-8-135
Supervised multivariate analysis of sequence groups to identify specificity determining residues
Abstract
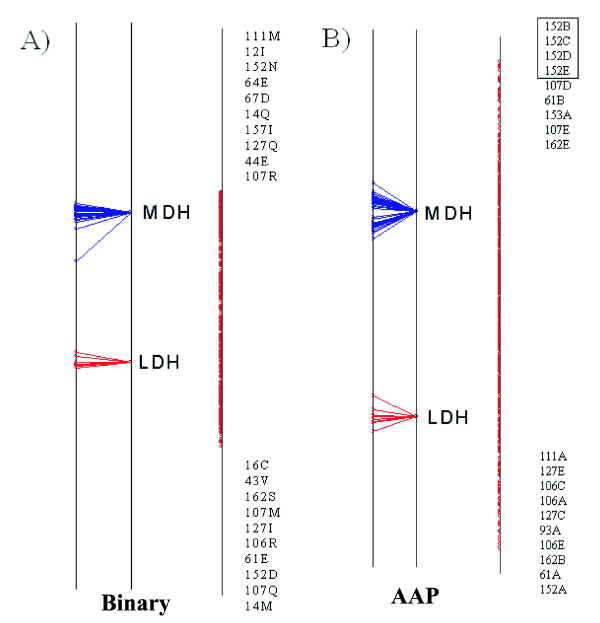
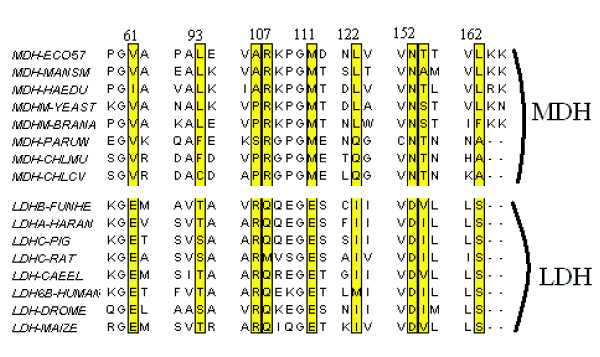
Background: Proteins that evolve from a common ancestor can change functionality over time, and it is important to be able identify residues that cause this change. In this paper we show how a supervised multivariate statistical method, Between Group Analysis (BGA), can be used to identify these residues from families of proteins with different substrate specifities using multiple sequence alignments.
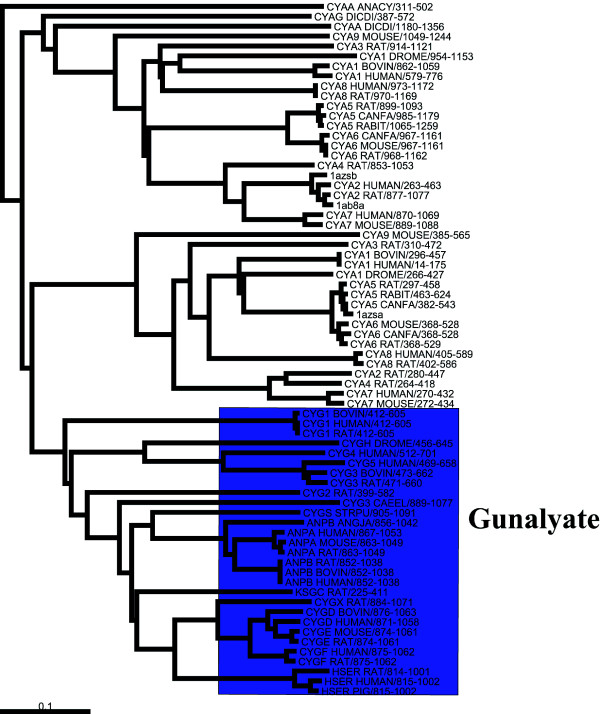
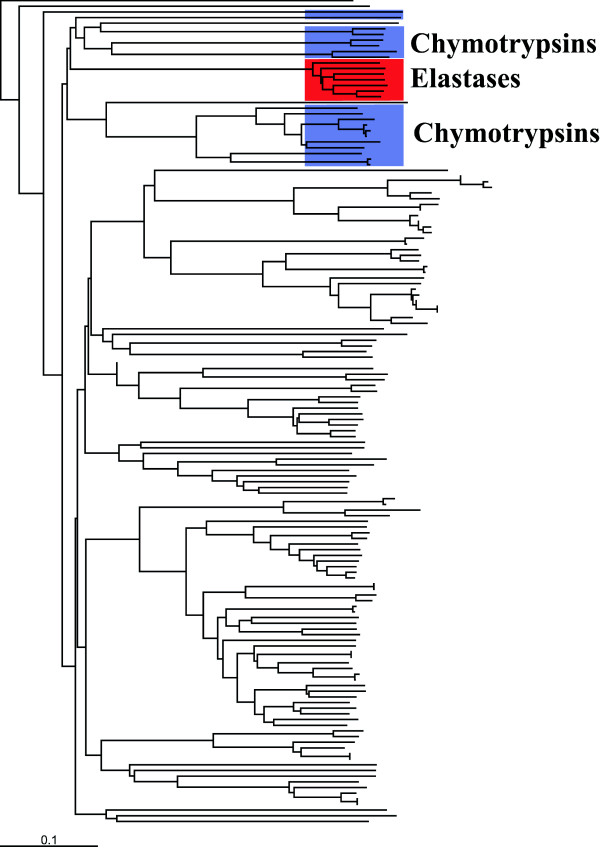
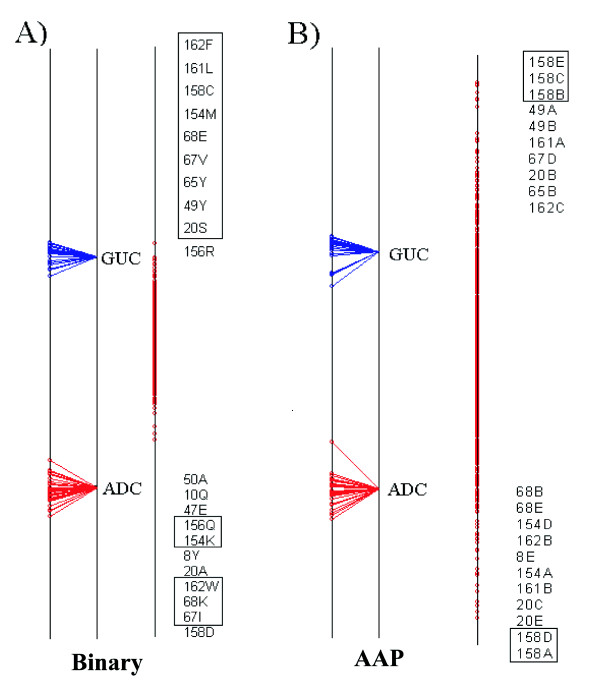
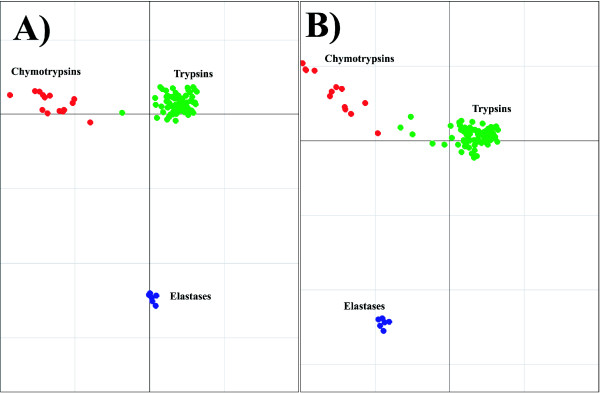
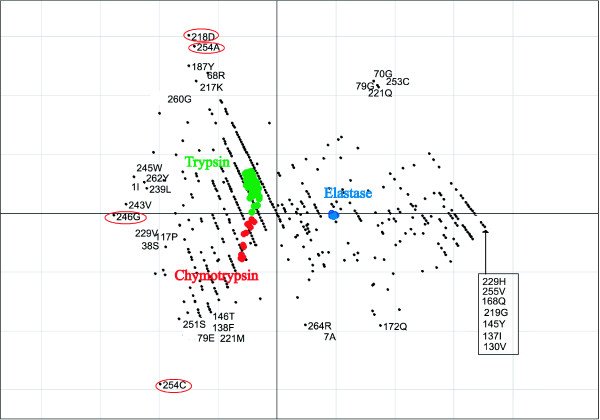
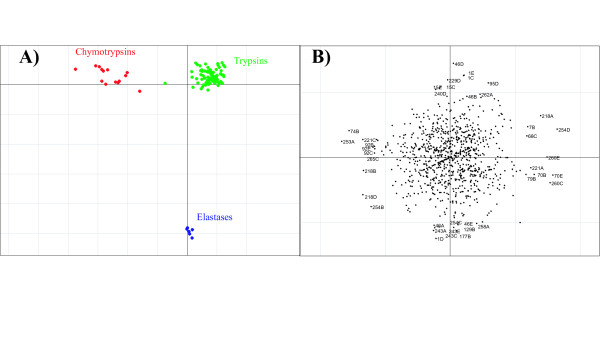
Results: We demonstrate the usefulness of this method on three different test cases. Two of these test cases, the Lactate/Malate dehydrogenase family and Nucleotidyl Cyclases, consist of two functional groups. The other family, Serine Proteases consists of three groups. BGA was used to analyse and visualise these three families using two different encoding schemes for the amino acids.
Conclusion: This overall combination of methods in this paper is powerful and flexible while being computationally very fast and simple. BGA is especially useful because it can be used to analyse any number of functional classes. In the examples we used in this paper, we have only used 2 or 3 classes for demonstration purposes but any number can be used and visualised.
Figures


References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
