

Pulmonary hypertension in sickle cell disease: relevance to children
- PMID: 17454785
- PMCID: PMC2065860
- DOI: 10.1080/08880010601185892
Pulmonary hypertension in sickle cell disease: relevance to children
Abstract
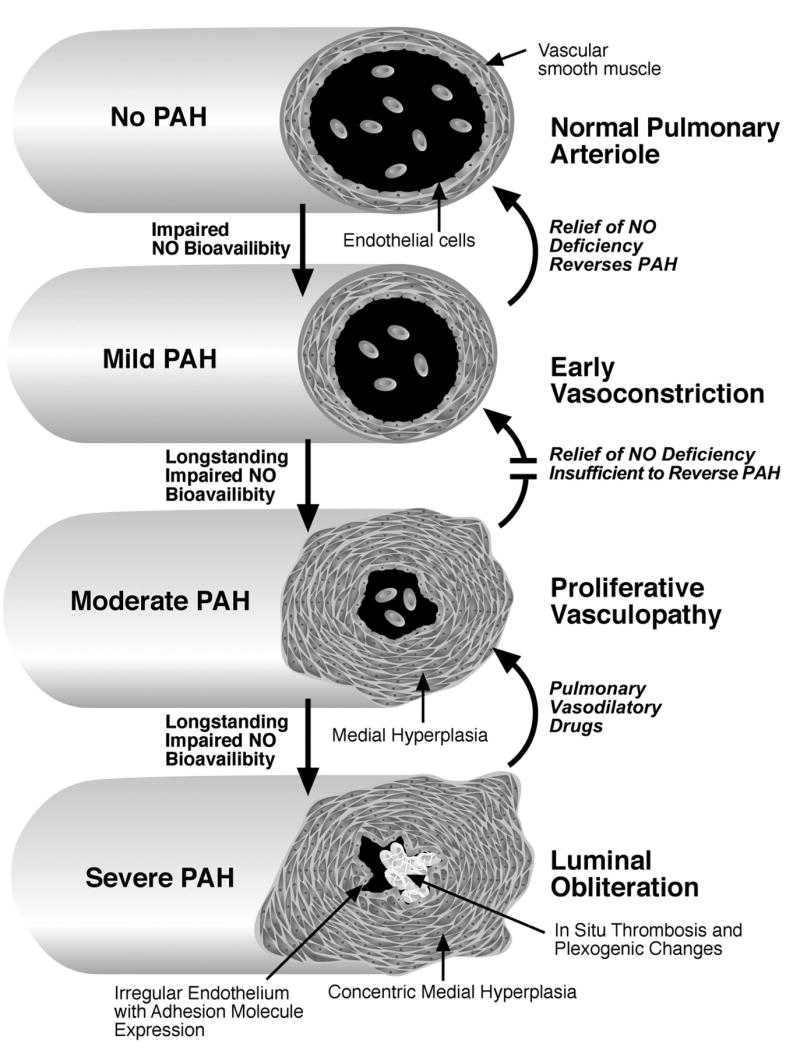
Pulmonary arterial hypertension (PAH), once considered a rare complication of sickle cell disease (SCD) and thalassemia, appears to be more common in adults with hemoglobinopathy than previously appreciated. On prospective screening of adults with SCD, approximately one-third of adults are found on echocardiography to have a tricuspid regurgitant jet velocity (TRV) of 2.5 m/s or higher, many of whom are asymptomatic. Dyspnea on exertion is the most common presenting symptom. This TRV abnormality is a marker for approximately 40% 3-year mortality in adults, and it is associated with laboratory values suggestive of more severe intravascular hemolysis. Release of hemoglobin and arginase from lysed red cells causes scavenging of nitric oxide (NO) and catabolism of L-arginine, the obligate substrate for NO synthase. The resulting impairment in NO bioavailability is associated with pulmonary vasoconstriction, endothelial dysfunction, thrombosis, and eventual development of plexogenic arterial lesions, the histological hallmark of all forms of PAH. Undoubtedly, additional pathophysiological mechanisms will also play a role in its multifactorial pathogenesis. Early data from children with SCD indicate a similar prevalence of elevated TRV, but the prognostic implications of this remain to be established. Individual patient diagnosis of PAH requires confirmation by right heart catheterization studies and individualized management. Hemolysis-associated PAH with impairments in NO bioavailability is being identified in thalassemia and other hemolytic disorders, and may be a general consequence of long-standing, severe intravascular hemolytic anemia.
Figures
Similar articles
-
Elevation of tricuspid regurgitant jet velocity, a marker for pulmonary hypertension in children with sickle cell disease.Pediatr Blood Cancer. 2006 Dec;47(7):907-13. doi: 10.1002/pbc.20791. Pediatr Blood Cancer. 2006. PMID: 16496290
-
Elevated tricuspid regurgitation velocity in congenital hemolytic anemias: Prevalence and laboratory correlates.Pediatr Blood Cancer. 2019 Jul;66(7):e27717. doi: 10.1002/pbc.27717. Epub 2019 Mar 25. Pediatr Blood Cancer. 2019. PMID: 30907497 Clinical Trial.
-
Platelet activation in patients with sickle disease, hemolysis-associated pulmonary hypertension, and nitric oxide scavenging by cell-free hemoglobin.Blood. 2007 Sep 15;110(6):2166-72. doi: 10.1182/blood-2006-12-061697. Epub 2007 May 29. Blood. 2007. PMID: 17536019 Free PMC article.
-
Pulmonary hypertension in sickle cell disease.Clin Adv Hematol Oncol. 2007 Aug;5(8):645-53, 585. Clin Adv Hematol Oncol. 2007. PMID: 17982405 Review.
-
Cardiomyopathy With Restrictive Physiology in Sickle Cell Disease.JACC Cardiovasc Imaging. 2016 Mar;9(3):243-52. doi: 10.1016/j.jcmg.2015.05.013. Epub 2016 Feb 17. JACC Cardiovasc Imaging. 2016. PMID: 26897687 Free PMC article. Review.
Cited by
-
The different facets of sickle cell disease-related pulmonary hypertension.Curr Opin Pulm Med. 2021 Sep 1;27(5):319-328. doi: 10.1097/MCP.0000000000000795. Curr Opin Pulm Med. 2021. PMID: 34224433 Free PMC article. Review.
-
Prospective evaluation of haemoglobin oxygen saturation at rest and after exercise in paediatric sickle cell disease patients.Br J Haematol. 2009 Nov;147(3):352-9. doi: 10.1111/j.1365-2141.2009.07854.x. Epub 2009 Aug 19. Br J Haematol. 2009. PMID: 19694721 Free PMC article.
-
TRV: a physiological biomarker in sickle cell disease.Pediatr Blood Cancer. 2012 Jun;58(6):831-2. doi: 10.1002/pbc.23399. Epub 2011 Dec 16. Pediatr Blood Cancer. 2012. PMID: 22180092 Free PMC article. No abstract available.
-
Hemodynamic predictors of mortality in adults with sickle cell disease.Am J Respir Crit Care Med. 2013 Apr 15;187(8):840-7. doi: 10.1164/rccm.201207-1222OC. Am J Respir Crit Care Med. 2013. PMID: 23348978 Free PMC article.
-
Pulmonary hypertension associated with chronic hemolytic anemia and other blood disorders.Clin Chest Med. 2013 Dec;34(4):739-52. doi: 10.1016/j.ccm.2013.08.006. Epub 2013 Oct 17. Clin Chest Med. 2013. PMID: 24267302 Free PMC article. Review.
References
-
- Moser KM, Shea JG. The relationship between pulmonary infarction, cor pulmonale and the sickle states. Am J Med. 1957;22:561–579. - PubMed
-
- Koren A, Garty I, Antonelli D, et al. Right ventricular cardiac dysfunction in beta-thalassemia major. Am J Dis Child. 1987;141:93–96. - PubMed
-
- Castro O, Hoque M, Brown BD. Pulmonary hypertension in sickle cell disease: cardiac catheterization results and survival. Blood. 2003;101:1257–1261. - PubMed
-
- Collins FS, Orringer EP. Pulmonary hypertension and cor pulmonale in the sickle hemoglobinopathies. Am J Med. 1982;73:814–821. - PubMed
-
- Sutton LL, Castro O, Cross DJ, et al. Pulmonary hypertension in sickle cell disease. Am J Cardiol. 1994;74:626–628. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical