

Is multiple-sequence alignment required for accurate inference of phylogeny?
- PMID: 17454975
- PMCID: PMC7107264
- DOI: 10.1080/10635150701294741
Is multiple-sequence alignment required for accurate inference of phylogeny?
Abstract
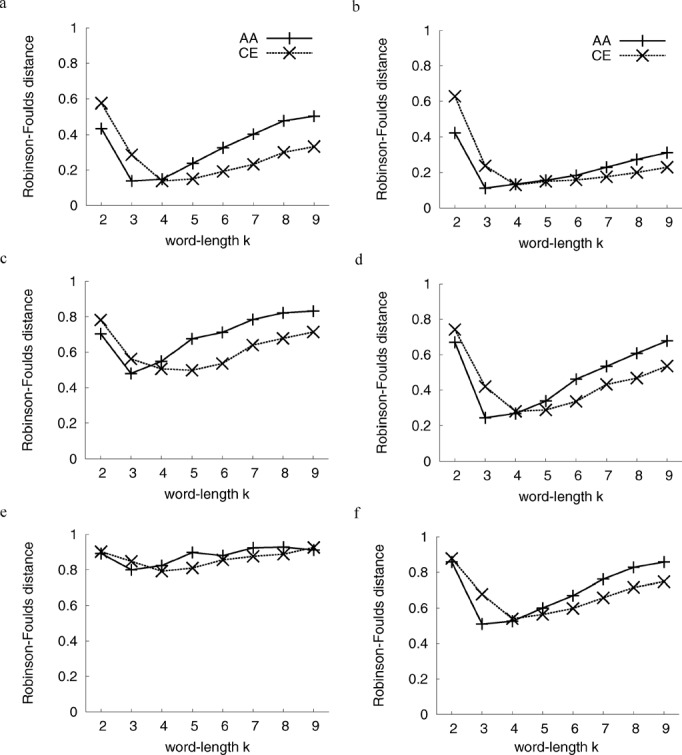
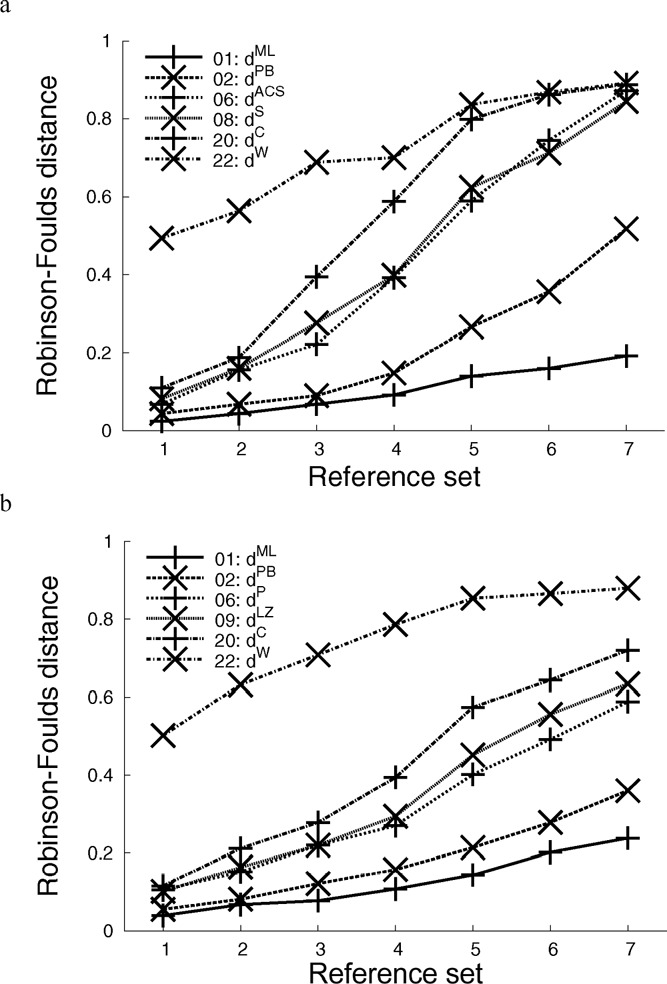
The process of inferring phylogenetic trees from molecular sequences almost always starts with a multiple alignment of these sequences but can also be based on methods that do not involve multiple sequence alignment. Very little is known about the accuracy with which such alignment-free methods recover the correct phylogeny or about the potential for increasing their accuracy. We conducted a large-scale comparison of ten alignment-free methods, among them one new approach that does not calculate distances and a faster variant of our pattern-based approach; all distance-based alignment-free methods are freely available from http://www.bioinformatics.org.au (as Python package decaf+py). We show that most methods exhibit a higher overall reconstruction accuracy in the presence of high among-site rate variation. Under all conditions that we considered, variants of the pattern-based approach were significantly better than the other alignment-free methods. The new pattern-based variant achieved a speed-up of an order of magnitude in the distance calculation step, accompanied by a small loss of tree reconstruction accuracy. A method of Bayesian inference from k-mers did not improve on classical alignment-free (and distance-based) methods but may still offer other advantages due to its Bayesian nature. We found the optimal word length k of word-based methods to be stable across various data sets, and we provide parameter ranges for two different alphabets. The influence of these alphabets was analyzed to reveal a trade-off in reconstruction accuracy between long and short branches. We have mapped the phylogenetic accuracy for many alignment-free methods, among them several recently introduced ones, and increased our understanding of their behavior in response to biologically important parameters. In all experiments, the pattern-based approach emerged as superior, at the expense of higher resource consumption. Nonetheless, no alignment-free method that we examined recovers the correct phylogeny as accurately as does an approach based on maximum-likelihood distance estimates of multiply aligned sequences.
Figures
References
-
- Beiko R. G., Chan C. X., Ragan M. A. A word-oriented approach to alignment validation. Bioinformatics. 2005;21:2230–2239. - PubMed
-
- Beiko R. G., Keith J. M., Harlow T. J., Ragan M. A. Searching for convergence in phylogenetic Markov chain Monte Carlo. Syst. Biol. 2006;55:553–565. - PubMed
-
- Castresana J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 2000;17:540–552. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
