

Differential regulation of AMPA receptor trafficking by neurabin-targeted synaptic protein phosphatase-1 in synaptic transmission and long-term depression in hippocampus
- PMID: 17460080
- PMCID: PMC6672995
- DOI: 10.1523/JNEUROSCI.5365-06.2007
Differential regulation of AMPA receptor trafficking by neurabin-targeted synaptic protein phosphatase-1 in synaptic transmission and long-term depression in hippocampus
Abstract
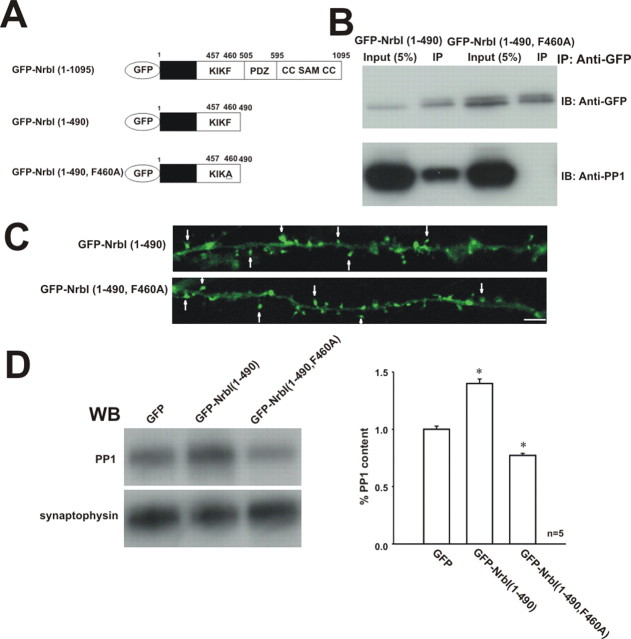
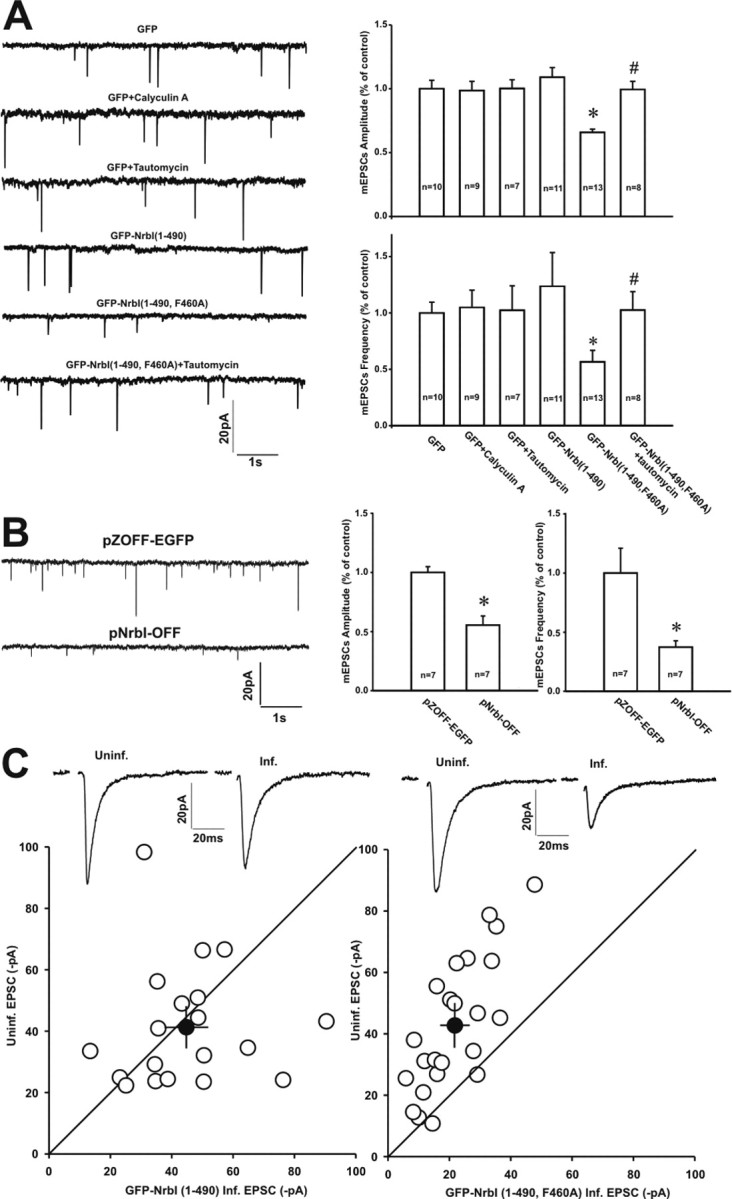
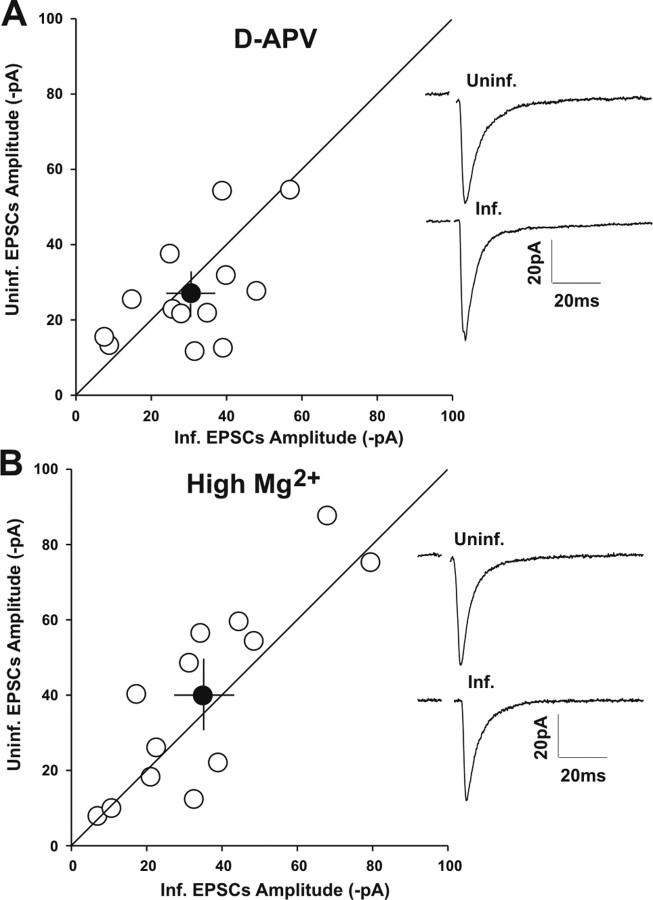
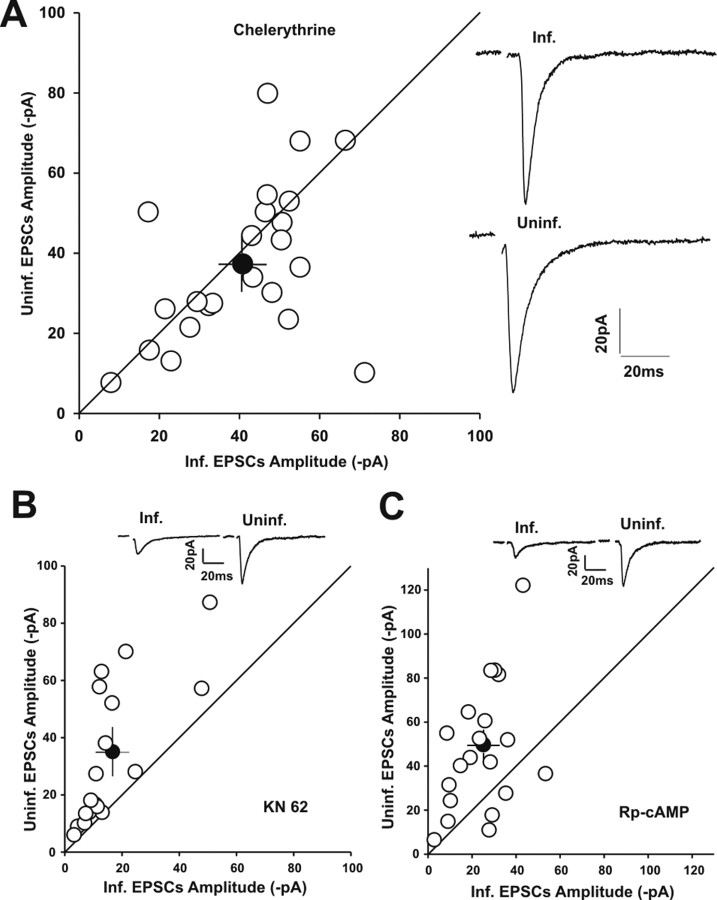
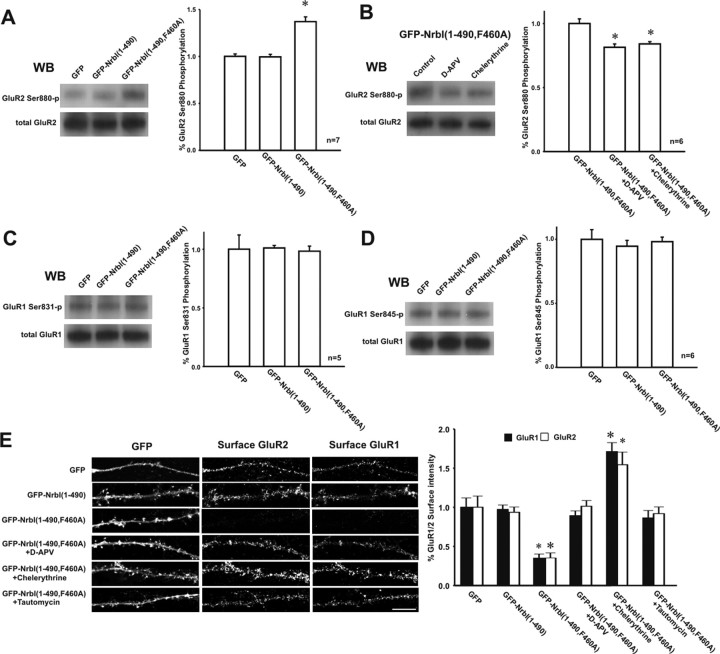
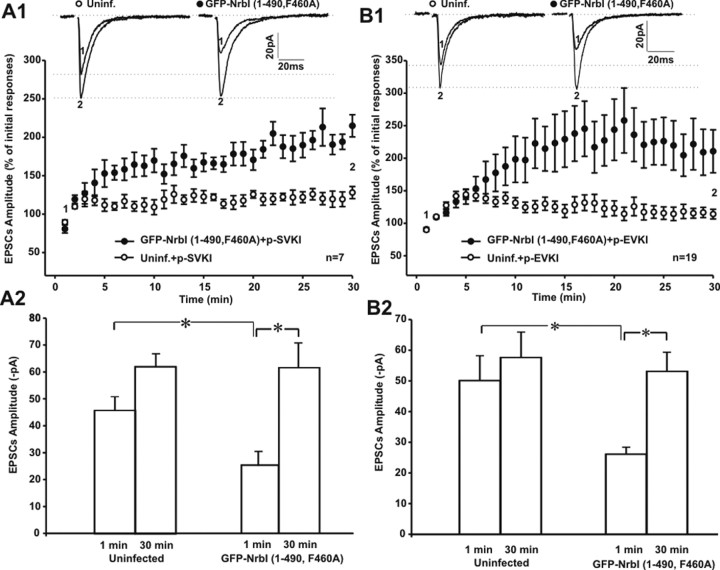
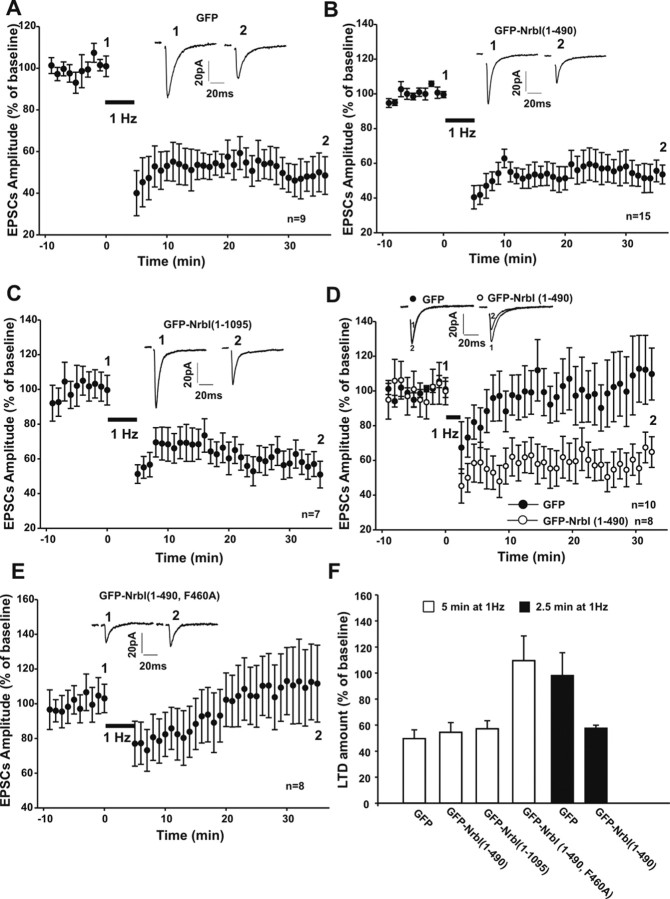
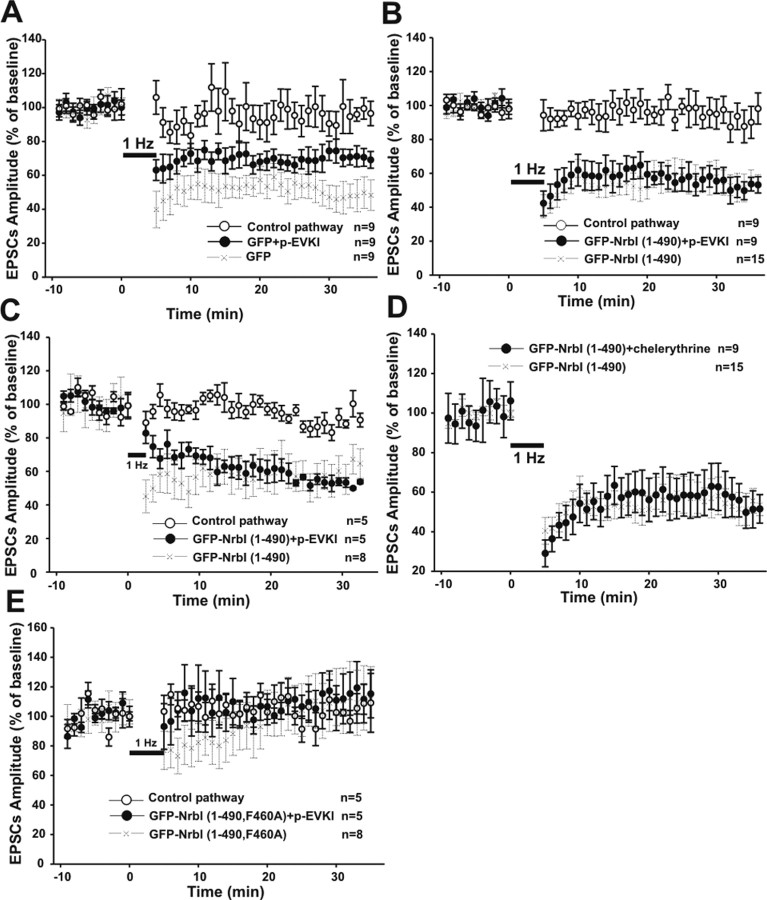
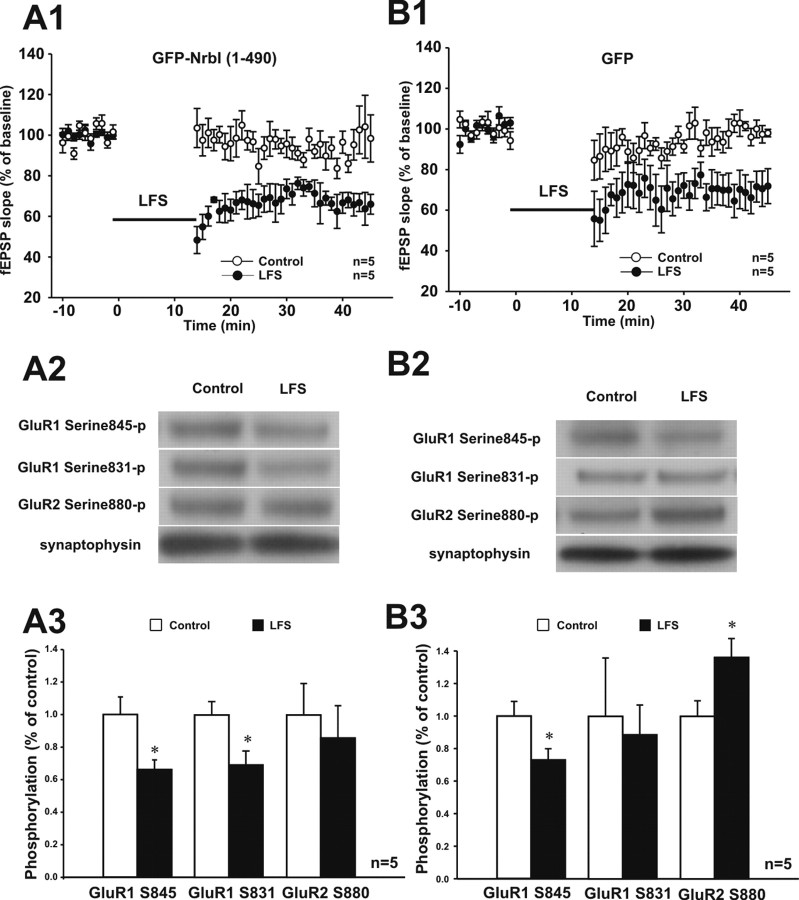
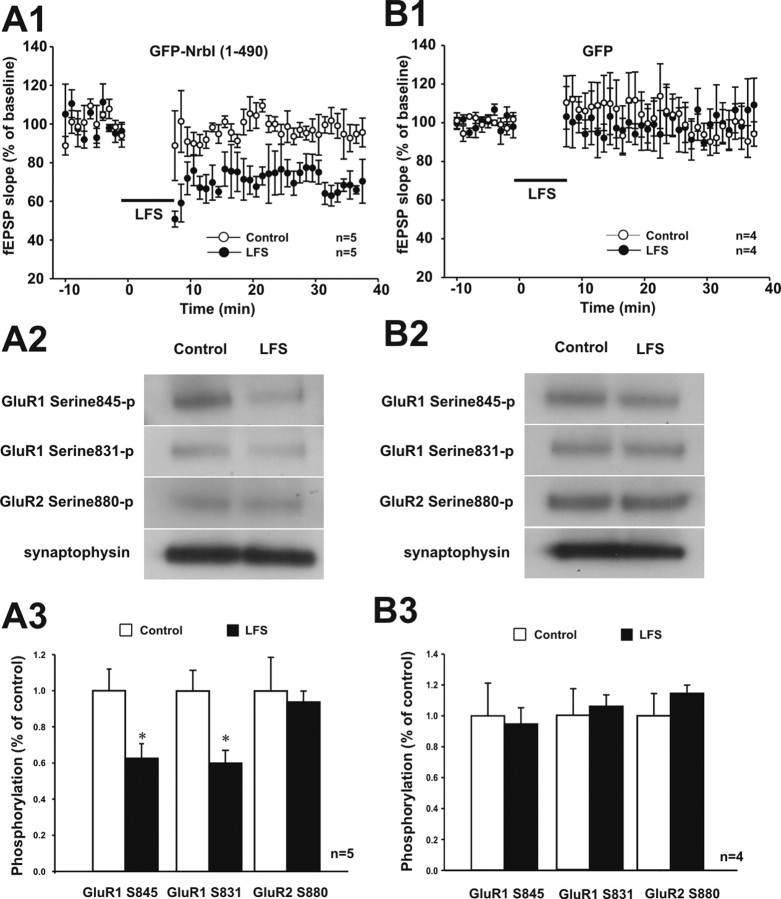
Filamentous actin binding protein neurabin I (NrbI) targets protein phosphatase-1 (PP1) to specific postsynaptic microdomains, exerting critical control over AMPA receptor (AMPAR)-mediated synaptic transmission. NrbI-targeted synaptic PP1, which promotes synaptic depression upon long-term depression (LTD) stimuli, serves to prevent synaptic depression under basal conditions. The present studies investigate this opposite regulation of AMPAR trafficking during basal synaptic transmission and LTD by expressing NrbI or NrbI mutant, which is defective in PP1 binding, in hippocampal slice or neuron cultures. We find that expression of the NrbI mutant to interfere with PP1 targeting dramatically reduces basal synaptic transmission, which is correlated with the reduction in surface expression of AMPA subtype glutamate receptor (GluR) 1 and GluR2 subunits. Biochemical analysis demonstrates that the NrbI mutant selectively increases the phosphorylation of GluR2 at C-terminal consensus PKC site, serine 880, which is known to favor GluR2 interaction with PDZ (postsynaptic density 95/Discs large/zona occludens 1) protein PICK1 (protein interacting with C kinase-1). Inhibition of PKC activity or GluR2-PICK1 interaction completely reverses the synaptic depression in neurons expressing the NrbI mutant, suggesting that NrbI-targeted synaptic PP1 stabilizes the basal transmission by negatively controlling PKC phosphorylation of GluR2 and the subsequent PICK1-mediated decrease in GluR2-containing AMPAR surface expression. Distinct from basal transmission, blocking GluR2-PICK1 interaction or PKC activity produces minimal effects on LTD in NrbI-expressing neurons. Instead, NrbI-targeted PP1 facilitates LTD by dephosphorylating GluR1 at both serine 845 and serine 831, with GluR2 serine 880 phosphorylation unaltered. Our studies thus elucidate that NrbI-targeted PP1, in response to distinct synaptic activities, regulates the synaptic trafficking of specific AMPAR subunits.
Figures
References
-
- Allen PB, Zachariou V, Svenningsson P, Lepore AC, Centonze D, Costa C, Rossi S, Bender G, Chen G, Feng J, Snyder GL, Bernardi G, Nestler EJ, Yan Z, Calabresi P, Greengard P. Distinct roles for spinophilin and neurabin in dopamine-mediated plasticity. Neuroscience. 2006;140:897–911. - PubMed
-
- Boehm J, Kang MG, Johnson RC, Esteban J, Huganir RL, Malinow R. Synaptic incorporation of AMPA receptors during LTP is controlled by a PKC phosphorylation site on GluR1. Neuron. 2006;51:213–225. - PubMed
-
- Bornancin F, Parker PJ. Phosphorylation of protein kinase C-alpha on serine 657 controls the accumulation of active enzyme and contributes to its phosphatase-resistant state. J Biol Chem. 1997;272:3544–3549. - PubMed
-
- Bredt DS, Nicoll RA. AMPA receptor trafficking at excitatory synapses. Neuron. 2003;40:361–379. - PubMed
-
- Carroll RC, Nicoll RA, Malenka RC. Effects of PKA and PKC on miniature excitatory postsynaptic currents in CA1 pyramidal cells. J Neurophysiol. 1998;80:2797–2800. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases