

Enzymatic analysis of a rhomboid intramembrane protease implicates transmembrane helix 5 as the lateral substrate gate
- PMID: 17463085
- PMCID: PMC1895938
- DOI: 10.1073/pnas.0700814104
Enzymatic analysis of a rhomboid intramembrane protease implicates transmembrane helix 5 as the lateral substrate gate
Abstract
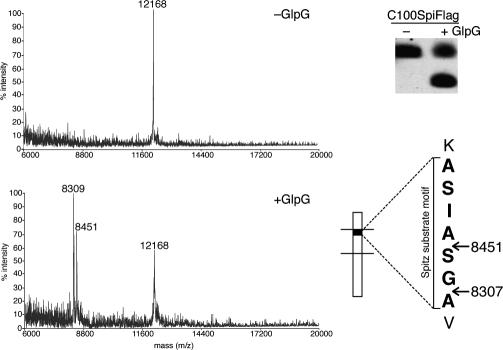
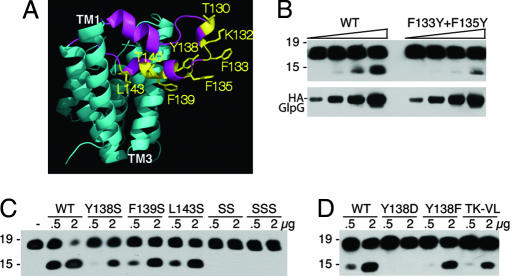
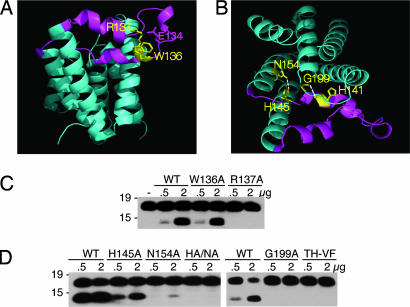
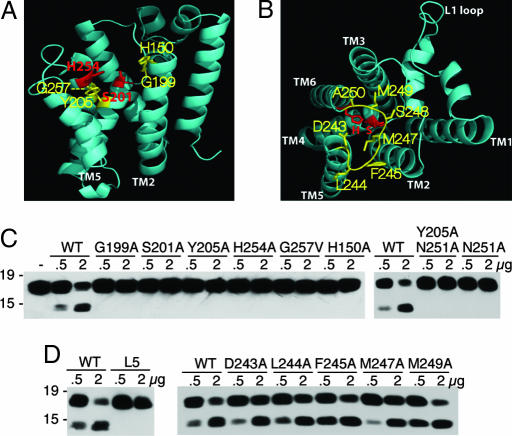
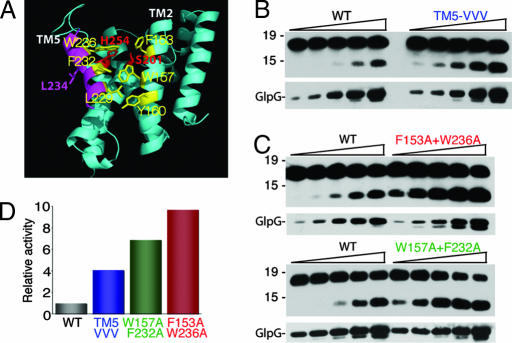
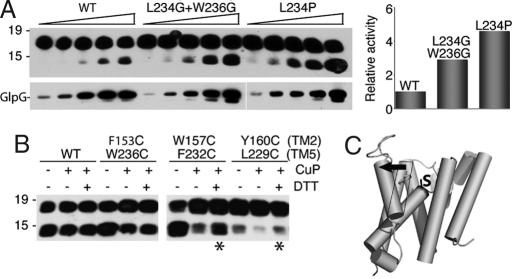
Intramembrane proteolysis is a core regulatory mechanism of cells that raises a biochemical paradox of how hydrolysis of peptide bonds is accomplished within the normally hydrophobic environment of the membrane. Recent high-resolution crystal structures have revealed that rhomboid proteases contain a catalytic serine recessed into the plane of the membrane, within a hydrophilic cavity that opens to the extracellular face, but protected laterally from membrane lipids by a ring of transmembrane segments. This architecture poses questions about how substrates enter the internal active site laterally from membrane lipid. Because structures are static glimpses of a dynamic enzyme, we have taken a structure-function approach analyzing >40 engineered variants to identify the gating mechanism used by rhomboid proteases. Importantly, our analyses were conducted with a substrate that we show is cleaved at two intramembrane sites within the previously defined Spitz substrate motif. Engineered mutants in the L1 loop and active-site region of the GlpG rhomboid protease suggest an important structural, rather than dynamic, gating function for the L1 loop that was first proposed to be the substrate gate. Conversely, three classes of mutations that promote transmembrane helix 5 displacement away from the protease core dramatically enhanced enzyme activity 4- to 10-fold. Our functional analyses have identified transmembrane helix 5 movement to gate lateral substrate entry as a rate-limiting step in intramembrane proteolysis. Moreover, our mutagenesis also underscores the importance of other residue interactions within the enzyme that warrant further scrutiny.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Comment in
-
From rhomboid function to structure and back again.Proc Natl Acad Sci U S A. 2007 May 15;104(20):8199-200. doi: 10.1073/pnas.0702745104. Epub 2007 May 9. Proc Natl Acad Sci U S A. 2007. PMID: 17494772 Free PMC article. No abstract available.
References
-
- Brown MS, Ye J, Rawson RB, Goldstein JL. Cell. 2000;100:391–398. - PubMed
-
- Wolfe MS, Kopan R. Science. 2004;305:1119–1123. - PubMed
-
- Urban S. Genes Dev. 2006;20:3054–3068. - PubMed
-
- Takasugi N, Tomita T, Hayashi I, Tsuruoka M, Niimura M, Takahashi Y, Thinakaran G, Iwatsubo T. Nature. 2003;422:438–441. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
